Team:NEU CHINA/Result

To enable engineered bacteria to successfully detect the coronavirus spike protein, the original Fe (III) sensitive domain Trp34-Glu64, an extracellular part of sensor protein PmrB, was replaced by the core binding domain of different coronavirus receptors. By this way, PmrB could respond to the stimulation of coronavirus spike protein and activated the downstream intracellular signal factor. It is well known that ACE2 is the receptor for SARS-CoV-2, DPP4 is the receptor for MERS-CoV, and hAPN is the receptor for HCoV-229E. To determine the effectiveness of multivirus detection system, SARS-CoV-2, MERS-CoV and HCoV-229E were used as examples. Therefore, gene fragments encoding ACE2, DPP4 and hAPN were cloned into plasmids to replace Trp34-Glu64 domain of PmrB, respectively.

Fig. 1 A: The plasmid was constructed to detect SARS-CoV-2’s spike protein. B: The plasmid was constructed to detect MERS-CoV’s spike protein. C: The plasmid was constructed to detect HCoV-229E’s spike protein.
A segment of peptide that can bind to the receptor in the spike protein of SARS-CoV-2, MERS-CoV or HCoV-229E was expressed and extracted, respectively. These peptides were used to simulate the environmental viruses, confirming the effectiveness of multivirus detection system.
 Fig. 2 Receptor binding domain of SARS-CoV-2 and core binding domain of ACE2.
Fig. 2 Receptor binding domain of SARS-CoV-2 and core binding domain of ACE2. Fig. 3 Receptor binding domain of MERS-CoV and core binding domain of DPP4.
Fig. 3 Receptor binding domain of MERS-CoV and core binding domain of DPP4. Fig. 4 Receptor binding domain of HCoV-229E and core binding domain of hAPN.
Fig. 4 Receptor binding domain of HCoV-229E and core binding domain of hAPN.A series of experiments was performed to examine the effectiveness of this system. IPTG (1mM) was used to induce the expression of PmrB and PmrA. Meanwhile, 34ng/𝝁l spike peptide of MERS-CoV, 34ng/𝝁l spike peptide of HCoV-229E, or 38ng/𝝁l spike peptide of SARS-CoV-2 was added into the culture medium, respectively. Once the engineered bacteria receive stimulation from the spike peptide, the sensor PmrB will autophosphorvlate and enable PmrA to activate the PmrC promoter, thus inducing the expression of reporter gene EGFP. Fluorescence intensity released by the bacteria under different circumstances was measured by enzyme-labeled instrument. Results indicated that the fluorescence intensity of the experimental groups (IPTG+S Pr) is all higher than the control (Ctrl) and IPTG groups (p<0.05). Generally speaking, the three systems all work.

Fig. 5 Fluorescence intensity of EGFP. Values represent the mean ± SEM. **p <0.01 versus control and ***p <0.001 versus control. Ctrl: Detection bacteria. IPTG: Detection bacteria + IPTG. IPTG+S Pr: Detection bacteria + IPTG+ S protein.
We set different IPTG concentration gradients to induce protein expression of the engineered bacteria, to find out the optimal amount for IPTG treatment. Under the concentration of IPTG in 10-4 mmol/L, the detection system’s fluorescence intensity is significantly higher than the control.
 Fig. 6 The fluorescence intensity of engineered bacteria with PmrCAB(ACE2).
Fig. 6 The fluorescence intensity of engineered bacteria with PmrCAB(ACE2). Fig. 7 The fluorescence intensity of engineered bacteria with PmrCAB(DPP4).
Fig. 7 The fluorescence intensity of engineered bacteria with PmrCAB(DPP4). Fig. 8 The fluorescence intensity of engineered bacteria with PmrCAB(hAPN).
Fig. 8 The fluorescence intensity of engineered bacteria with PmrCAB(hAPN).The results suggested that engineered bacteria induced by IPTG could sense the signal from the virus and export the report signal. As we expected, the PmrCAB system is feasible for coronavirus detection.
It is well known that quorum sensing (QS) system allows the first activated bacteria to transfer the activation signal from individual one to the population. Thus, to improve the sensitivity of multivirus detection system, quorum sensing system was applied. The plasmids were constructed using pETDuet-1 and pACYCDuet-1 as the basic framework. The QS gene LuxI/LuxR was added behind the PmrC. LuxI catalyzes the synthesis of the signaling molecule AHL, which binds to LuxR to form a complex that activates the PLux_HS promoter to express downstream genes.
In this way, the stimulation signal received by PmrCAB system will be transferred to the QS system immediately, then spreading to other bacteria by AHL. Therefore, a number of bacteria will be activated by the QS system, making the multivirus detection system detect extremely low virus particle number.
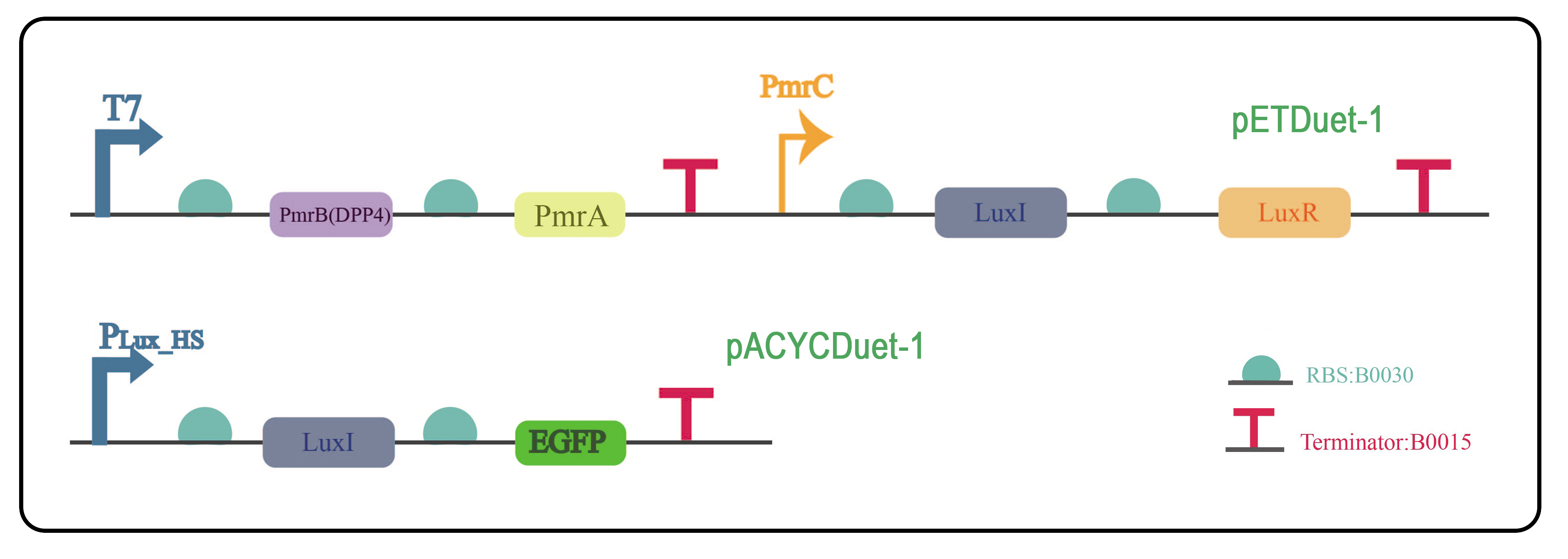
Fig. 9 The two plasmids were constructed with pETDuet-1 and pACYCDuet-1 as the vectors, and were co-transformed to E.coli BL21(DE3) to verify the quorum sensing system.
To verify whether the quorum sensing system is effective, the following experiments were conducted.
IPTG was added to induce the expression of PmrB and PmrA, followed by 9.4 ng/𝝁l of MERS-CoV spike peptide. The fluorescence intensity was measured every hour. As shown in Fig.10 and Fig. 11, we observed the three groups of bacteria with fluorescence microscope after the induction. Strong green fluorescence can be obviously noticed at the group of PmrCAB+QS. And other two groups have less fluorescent signals. And the fluorescence intensity reached a high level when the engineered bacteria being treated with 10-4 mmol/L IPTG and spike peptide for 4 hours.

Fig. 10 Fluorescence microscopy photos of detection bacteria. A: Engineered bacteria with empty vector. B: Engineered bacteria with PmrCAB. C: Engineered bacteria with PmrCAB + QS.
 Fig. 11 The fluorescence intensity of engineered bacteria without quorum sensing system and with quorum sensing.
Fig. 11 The fluorescence intensity of engineered bacteria without quorum sensing system and with quorum sensing.It confirms that quorum sensing system can solve the false negative problem caused by low virus concentration in the environment to a certain extent.
To determine the specific threshold of AHL, the following plasmids as shown in Fig. 12 were constructed.
Fig. 12 The plasmid was constructed with pACYCDuet-1 as the vector to explore the concentration threshold of AHL.The extracellular release of AHL from bacteria under natural conditions was simulated by artificially adding AHL. A concentration gradient of AHL (0 𝝁mol/L to 13 𝝁mol/L) was set. When bacteria grew into logarithmic phase, different concentrations of AHL were added into the culture medium. AHL combined with endogenous LuxR could activate the PLux_HS promoter, to induce the expression of downstream genes, finally induce EGFP expression. The fluorescence intensity was then measured. As shown in Fig. 13, the fluorescence intensity in engineered bacteria with QS was enhanced at a AHL-dose dependent manner and step into the plateau period when AHL reached 9 μmol/L.
 Fig. 13 The fluorescence intensity of engineered bacteria under different concentrations of AHL.
Fig. 13 The fluorescence intensity of engineered bacteria under different concentrations of AHL.