PART COLLECTION
ContributionImprovement

| Part No. | Name | Description | Type |
|---|---|---|---|
| BBa_K3728000 | Tol2 transposase (Oryzias latipes) - Tpase | Transposase in Tol2 transposon system | Basic part - coding |
| BBa_K3728002 | pTol2: a Tol2 vector - the Tol2 transposable element | Tol2 vector in Tol2 transposon system | Plasmid backbone |
| BBa_K3728003 | J04450/pTol2 - BioBrick compatible Tol2 transposable vector | BioBrick compatible vector | Standardized composite part |
| BBa_K3728004 | KanR/pTol2 | Kanamycin resistance cassette on Tol2 vector | Antibiotic resistance |
| BBa_K3728005 | ldhp-GFP-Tr/pTol2 | Constitutive GFP-expressing Tol2 vector | Reporter (GREEN FLUORESCENCE) |
| BBa_K3728006 | ldhp-RFP-Tr/pTol2 | Constitutive RFP-expressing Tol2 vector | Reporter (RED) |
| BBa_K3728007 | ldhp-amilCP-Tr/pTol2 | Constitutive amilCP-expressing Tol2 vector | Reporter (BLUE) |
| BBa_K3728008 | ldhp-Phi29 DNA pol-Tr/pTol2 | Constitutive phi29 DNA polymerase-expressing Tol2 vector | Reporter (for RCA) |
| BBa_K3728009 | pSB6K1 - a pBR322-based vector with Kanamycin resistance | A plasmid backbone with pBR322 ori and KanR | Plasmid backbone |
| BBa_K3728010 | pSB6C1 - a pBR322-based vector with Chloramphenicol resistance | A plasmid backbone with pBR322 ori and CmR | Plasmid backbone |
| BBa_K3728013 | ldhp-RFP-Tr/pSB6C1 | Constitutive RFP-expressing pBR322-based vector | Reporter (RED) |
This year we focused on developing a Salmonella diagnosis tool. For safety issues such as Salmonella at Biosafety Level 2, transposase activity involved in Gene Drive, etc., we’ve built up a simplified and standardized cell-free in vitro transcription-translation (TXTL) system and phage/Salmonella engineering toolkit that will benefit iGEM projects in the future.
The BioBrick Parts we made and collected as an Engineering Toolkit were categorized by bacteriophage genome engineering based on in vitro Tol2 transposon system and Salmonella transformation based on pBR322-based vectors.
Based on Tol2 transposon system, we created Tol2 transposase [BBa_K3728000] and a vector of pTol2 composed of the minimal Tol2 transposable elements [BBa_K3728002]. The pTol2 was made compatible with general BioBrick Assembly Rule by connecting BBa_J04450 and deleting conflict restriction enzyme sites [BBa_K3728003]. Moreover, we demonstrated the application of TXTL and Tol2 systems [BBa_K3728004-8] for in vitro engineering a Salmonella phage genome and synthesizing infectious phage by in vitro packaging.
In addition, when working on multi-drug resistant Salmonella studies, we improved an existing part of pSB6A1 vector by exchanging antibiotic resistance genes [BBa_K3728009-10, 13] that can be used in the Salmonella spp. studies.
Our construction and characterization of parts will represent our efforts and achievement in Part Collection.
Tol2 transposon system is highly used in zebrafish transgenesis. The transposase protein (TPase) is from the Medaka fish (Oryzias latipes) aka Japanese rice fish, which catalyzes the transposition of the Tol2 elements through cut-and-paste mechanism. The minimal transposable Tol2 sequence (mTol2) contains 200-bp left arm and 150-bp right arm1. Up to 11kb DNA insert between Tol2 sequence can be integrated into the genome of nearly all vertebrates including zebrafish, frog, chicken, mouse, and human2.
A further application in synthetic biology was demonstrated by Jun Ni, et. al. 3, in which the recombinant TPase protein is fully functional in HeLa cell line and Zebrafish germline cells. In addition, the TPase can be expressed under T7 promoter in E. coli BL21 and purified with N-terminal 6xHis tag. The transposase is active in vitro and mediated the integration of DNA fragments between plasmids with Tol2 elements.
To engineer an isolated phage genome with unknown full genome sequence in our project, we want to create Tol2 transposon system. The system contains a HELPER (Tol2 tranposase, TPase), a DONOR (Tol2 mobile element) which becomes a vector to carry CARGO (phi29 DNA polymerase) jumping into a target (Salmonella phage genome). The engineered phage genome with recombinant phi29 DNA polymerase gene will be synthesized and packaged in TXTL reaction to acquire the infectious Salmonella phages.
Oryzias latipes Tol2 transposase (TPase) gene sequence was taken from UniProt database4 and optimized based on E. coli codon preference. The TPase gene was designed with 6xHis tag and a GS linker at N-terminus and synthesized by Integrated DNA Technologies, Inc. (IDT). The DNA fragment was cloned into pSB1C3 as a basic BioBrick part (TPase/pSB1C3, Part:BBa_K3376000). The part was checked by colony PCR and restriction enzymes and further confirmed by sequencing (Fig. 1).

Figure 1 |6xHis-GS-TPase/pSB1C3 construct check. DNAs were run electrophoresis on 1% agarose gel with 1kb marker. (a) 4 colonies were subject to PCR with TPase-specific forward and reverse primers (PCR product size: ~2000 bp). (b) The DNAs were extracted and digested by EcoRI and BamHI (2893, 870 and 235 bps).
His-tagged Tol2 transposase (TPase) was assembled with a T7 promoter and expressed in TXTL reaction with the bacterial cytoplasmic extracts prepared from IPTG-induced E. coli Rosetta 2(DE3) cells. The His-tagged proteins were further purified through Nickel column. The protein concentration was measured and analyzed on SDS-PAGE and Coomassie Blue Staining (Fig. 2). The protein was shown at around 70 kDa as the same size as the predicted TPase protein (664 a.a., 75 kDa). The Elution #4, #5 and #6 were collected and used for further studies.

Figure 2 |His-Tol2 transposase was expressed in TXTL and purified by Nickel column. 10 μg of protein lysates were analyzed by SDS-PAGE and Coomassie Blue Staining using 4–12% gradient gel (NuPAGE™, Thermo Fisher Scientific Inc.) Lane: (1) PageRuler™ Prestained Protein Ladder, (2) E. coli Rosetta 2(DE3) cell extracts (no DNA control), (3) total lysates in TXTL, (4) flow-through, (5) wash-through, (6) Elution #4, (7) Elution #5, (8) Elution #6, (9) Elution #7, (10) Elution #8, (11) Elution #9.
In vitro integration assay was used by Jun Ni, et al. to characterize the activity of purified recombinant Tol2 transposase (TPase) and the transposition of Tol2 mobile element3. We prepared the purified TPase from TXTL (Fig. 2) and performed PCR to generate KanR, ldhp-GFP-Tr and ldhp-amilCP-Tr (expressing blue color) DNA fragments flanked by 200-bp right and 150-bp left arms of pTol2. The mixtures of TPase, Tol2 mobile inserts and a target plasmid of pSB1C3 were incubated at 30°C for 2 hours. The resulting DNAs were cleaned up and subjected to transform E. coli DH5α competent cells. The colonies displaying kanamycin resistance, green fluorescence or blue color were counted as successful jumping to plasmids by active purified TPase. And the integration rate was calculated by comparing with chloramphenicol resistance or red colonies from pSB1C3 backbone carrying BBa_J04450 part (i.e., RFP coding device).

GFP/Tol2-integrated plasmid can transform E. coli to exhibit weak to strong green fluorescence in Fig. 3. Two plasmids of GFP-positive bacteria were extracted and checked by restriction enzymes. They are larger than pSB1C3 when single cut on the backbone by ApaLI (Fig. 4b). The schematic map of Fig. 4a showed the possible position of integration by a BamHI-cut on the insert and a ApaLI-cut on the backbone (Fig. 4c). The rate of successful integration was calculated by the ratio of numbers of KanR, GFP and BLUE colonies to CmR or RED colonies, respectively (Fig. 4d). The ratio was between 0.2% to 0.9%, of which data are consistent with the observation by Jun Ni, et al3. In sum, we can modify plasmid DNAs in vitro with an insert between Tol2 mobile elements (DONOR) and purified TPase enzymes (HELPER) from TXTL reaction.

Figure 3 |E. coli colonies on Cm agar plates were transformed by the mixture of GFP/Tol2 and pSB1C3 with TPase or without TPase as a control.

Figure 4 |Possible integration map and ratio. (a) Schematic maps showed the predicted integration sites. (b, c) pSB1C3::GFP/Tol2 Clone #1 (lane 1) and #2 (lane 2) or pSB1C3 as a control (lane 3) were cut by ApaLI on the backbone (b) or cut by ApaLI with a BamHI cut on the insert (c). DNA was analyzed by electrophoresis on 1% agarose gel with a 1kb marker. (d) The successful integration ratios are calculated by the numbers of colonies of pSB1C3::KanR/Tol2 on Kan agar plate divided by those of pSBC13 (CmR) on Cm agar plates or by the numbers of pSB1C3::GFP or pSB1C3::BLUE divided by colony numbers of pSB1C3 (RED) on Cm agar plates such as shown in Figure 3.
The genome of Salmonella phage #ST1 was extracted and engineered to carry phi29 DNA polymerase gene through the Tol2 transposon system.

T7-His-Tol2 transposase was expressed in TXTL using IPTG-induced E. coli Rosetta 2(DE3) extracts, followed by purification with Nickel column. The DNA fragment of Tol2 transposable element carrying ldhp-Phi29 DNA polymerase-Tr (Ф29/Tol2) was amplified by PCR. The extracted Salmonella phage genome and the Ф29/Tol2 DNA fragment were incubated with Tol2 transposase at 30°C for 2 hours. The recombinant phage genome was subjected to TXTL based on the work of Jonghyeon Shin5, where T7 phage genome can be replicated, synthesized, and assembled in a single cell-free reaction. The infectious phage of Salmonella phage #ST1::Ф29 made by TXTL using Salmonella extracts was tested on plaque assay with a culture of Salmonella on the LB agar plate. As shown in Fig. 5b, visible different sizes of plaques were formed on the agar plate, demonstrating infectious phages were produced in our TXTL system. As a control, the phage gDNA without TXTL reaction displayed no plaques (Fig. 5a), indicating the live phages from TXTL are not from the contaminated DNA in the process of genomic DNA extraction.

Figure 5 |The recombinant phage synthesis in TXTL. Plaques were formed on a lawn of Salmonella culture on the LB agar plate from TXTL reaction (b) compared to no plaques without TXTL reaction (a).
Dozens of plaques were screened by PCR with phi29 DNA polymerase gene-specific primers. A representative result on DNA gel electrophoresis was shown in Fig. 6, in which the successful Ф29/Tol2 insertion (Salmonella phage::Ф29 DNA polymerase, or #ST1::Ф29 for short) can be amplified by PCR with either Tol2 transposable element-specific primers or phi29 DNA polymerase-specific primers, compared to no PCR products from wild-type Salmonella phage #ST1, showing the success of our Salmonella phage engineering with phi29 DNA polymerase gene in in vitro Tol2 transposon system using TXTL-expressed Tol2 transposase.

Figure 6 |Salmonella genome were checked by PCR with Tol2 transposable element-specific primers (lanes 1, 2) or with phi29 DNA polymerase gene-specific primer set 1 (lanes 3, 4) or set 2 (lane 5, 6). The odd numbers refer to Salmonella phage::Ф29 DNA polymerase (#ST1::Ф29), and the even numbers refer to wild-type Salmonella phage #ST1. The gel electrophoresis was performed on a 1% agarose gel with a 1kb DNA ladder.
Phi29 (Φ29) DNA polymerase is an exceptional processive polymerase possessing highly isothermal amplification and strong strand displacement activities6,7. The polymerase has been increasingly used in rolling circle amplification (RCA) assay that make long ssDNA strands using an initial primer and a single-stranded circular DNA template in the buffer containing dNTPs and Mg2+. The large amounts of amplified DNAs can be quickly generated within 1 hour and easily be measured with fluorescent DNA binding dye (e.g., EvaGreen® Dye).

To engineer an isolated Salmonella phage genome with unknown full genome sequence, we want to use in vitro Tol2 transposon system with Tol2 transposase and Tol2 transposable element to make a recombinant phage genome carrying phi29 DNA polymerase gene.
To achieve that, we built up a Tol2 transposable vector of the BioBrick part of ldhp-Phi29 DNA pol-Tr/pTol2 consisting a constitutive promoter of ldhp and a His-tagged phi29 DNA polymerase gene with a double terminator. We verified the functionality of each basic part on the ldhp-Phi29 DNA pol-Tr/pTol2 in the following. Finally, we demonstrated a Salmonella phage engineered with this vector can be a useful Salmonella detection tool.
Minimal Tol2 transposable element (mTol2) has been characterized that is composed of 200-bp left arm and 150-bp right arm1. The 19-bp to 11-kbp DNA inserts between the arms can be excised and transposed efficiently by Tol2 transposase (TPase). Therefore, we’d like to make a BioBrick compatible vector based on Tol2 mobile element (pTol2), which can be further assembled through a BioBrick standard EcoRI-XbaI-SpeI-PstI rule.
We obtained the backbone of pBSII-SK-mTol2-MCS from Addgene (Plasmid #51817), which was given by Elly Tanaka8. We deleted restriction enzyme sites in MCS and generated novel BioBrick Prefix (EcoRI-NotI-XbaI) and BioBrick Suffix (SpeI-NotI-PstI) elements in the both ends by PCR. The resulting DNA plasmid backbone called pTol2 (Part:BBa_K3376002) was further assembled with the Part BBa_J04450 (i.e., the iGEM official standard insert on pSB1C3). The resulting J04450/pTol2 (Part:BBa_K3376003) was checked by PCR (Fig. 7a) and restriction enzymes (Fig. 7b) and also confirmed by sequencing.

Figure 7 |pTol2 and J04450/pTol2 constructs check. DNAs were run electrophoresis on 1% agarose gel with 1kb marker. (a) PCR producs of pTol2 (lane 1, 3429 bp) and BBa_J04450 (lane 2, 1112 bp). (b) 4 clones of J04450/pTol2 were checked by restriction enzymes (~ 3432 bp and ~1110 bp). Lanes 1-4 by EcoRI and SpeI. Lanes 5-8 by XbaI and PstI.
To engineer RCA reporter Salmonella phage, we requested Part:BBa_K3352001 (Φ29 DNA Polymerase with His-Tag and GS linker Sequence) from iGEM team TAS_Taipei. The part was assembled with ldhp promoter (Part:BBa_K3376000) and a double terminator (Part:BBa_B0015) onto the Tol2 mobile element vector (pTol2). The resulting composite part named ldhp-Phi29 DNA pol-Tr/pTol2 (Part:BBa_K3728008) was checked by colony PCR, restriction enzymes and sequencing (Fig. 8).

Figure 8 |ldhp-Phi29 DNA pol-Tr/pTol2 construct check. DNAs were run electrophoresis on 1% agarose gel with 1kb marker. (a) 4 colonies were subject to PCR with ldhp forward primer and a reverse primer in the end of Phi29 gene (PCR product size: 1965 bp). (b) The DNAs were extracted and digested by EcoRI and PstI (3573 and 1973 bps).
To test the TPase activity and Tol2 transposon system, we inserted a kanamycin resistance gene (KanR) cassette (Part:BBa_K3376004) and the reporters of ldhp-GFP-Tr(Part:BBa_K3376005), ldhp-RFP-Tr(Part:BBa_K3376006) and ldhp-amilCP-Tr(Part:BBa_K3376007) between the transposable elements on the pTol2 vector. ldhp is a constitutive and broad- host-range promoter, which was originally cloned and driving the lactate dehydrogenase gene in S. mutans. We have characterized the ldhp activities in S. mutans and E. coli in our project of iGEM 2020, as well as Salmonella and TXTL in this project of iGEM 2021. (for detail, go to check our (CONTRIBUTION page)
The GFP and RFP fluorescence intensities driven by ldhp on pTol2 vectors were measured at high level in TXTL reaction (Fig. 9). The strong GFP fluorescence can even be visualized by naked eyes under a Blue LED Illuminator. Compared the activities of ldhp to lac promoter (lacp), lacp is inhibited in TXTL because the extracts of E. coli Rosetta 2 (DE3) contains LacI repressor, which can be relieved by IPTG induction or using E. coli DH5α as extracts.

Figure 9 |Promoter activities on pTol2 vector in TXTL. GFP fluorescence was measured at Ex/Em = 500/530 nm using a microplate reader of BioTek Synergy H1. RFP was at Ex/Em = 586/611 nm. KanR/pTol2 in TXTX was set as a background control. AU means arbitrary unit. (a) ldhp-GFP-Tr/pTol2 activity in TXTL. The inset photo was captured under a blue LED light. (b) ldhp-RFP-Tr/pTol2 and J04450/pTol2 (i.e., lacp-RFP-Tr) in TXTL.
To characterize the function of recombinant phi29 DNA polymerase, TXTL cell-free system can solve the problem caused by the difficulty in bacterial transformation or without suitable plasmid vectors. We took the DNA of ldhp-Phi29 DNA pol-Tr/pTol2 mixed into TXTL reaction with Salmonella extracts. The recombinant His-tagged phi29 DNA polymerase protein was purified through Nickel column and analyzed by SDS-PAGE and Coomassie Blue Staining (Fig. 10). The isolated proteins in Elution #4 and #5 were at the size of around 70 kDa as predicted (His-phi29 DNA polymerase: 590 amino acids, 68 kDa) and collected for the following studies.

Figure 10 |His-phi29 DNA polymerase was expressed in TXTL using Salmonella extracts and purified by Nickel column. 5 μg of protein lysates were analyzed by SDS-PAGE and Coomassie Blue Staining using 4–12% gradient gel (NuPAGE™, Thermo Fisher Scientific Inc.) Lane: (1) PageRuler™ Prestained Protein Ladder, (2) Salmonella cell extracts (no DNA control), (3) total lysates in TXTL, (4) flow-through, (5) wash-through, (6) Elution #3, (7) Elution #4, (8) Elution #5, (9) Elution #6, (10) Elution #7, (11) Elution #8.
We performed RCA by mixing a circular ssDNA and primer in the buffer with our purified phi29 DNA polymerase (Φ29) from TXTL using Salmonella extracts or a commercial recombinant Φ29 from New England Biolabs Inc. (NEB) as a positive control. The Φ29 enzymes were diluted with various factors in the assay. After incubation at 30°C for 1 hour, the RCA products were stained with EvaGreen DNA dye and subjected to a microplate reader to measure signals at Ex/Em=500/530 nm. And the fold changes in fluorescence intensity were calculated by dividing the values from Φ29-untreated groups (as controls). As shown in Fig. 11, a 12-fold change was achieved with our Φ29, indicating the functionality and the activity of ldhp-Phi29 DNA pol-Tr/pTol2 that are comparable to the commercial NEB Φ29 enzymes. Moreover, the green fluorescence can readily be seen with a blue led light, even using a 4 times diluted Φ29, proving the super high processivity of phi29 DNA polymerase in DNA amplification.

Figure 11 |RCA assay using TXTL-expressed (MINGDAO) or commercial (NEB) phi29 DNA polymerase (Φ29). The commercial Φ29 was purchased from NEB with a defined activity by units. The control was set without Φ29 treatment. The concentration and dilutions of enzymes were, respectively, 4, 2, 1 μg/μl for MINGDAO Φ29 and 1, 0.5, 0.25 units for NEB Φ29. The EvaGreen DNA binding signals were read at Ex/Em=500/530 nm in BioTek Synergy H1 Microplate Reader.
An overnight culture of Salmonella Typhimurium LT2 (~109 cells/ml) were infected by the engineered reporter phage #ST1::Ф29 at MOI=0.1 to produce phi29 DNA polymerase. The lysates were collected after 2 hr or 4 hr of treatment, and then subjected to RCA test. The lysate of phage-infected Salmonella at 2 hr can induce strong RCA reaction (24-fold change) comparable to 2.5U of a commercial phi29 DNA polymerase (NEB) in Fig. 12. Surprisingly, the lysate at 4 hr can not trigger any signal in RCA, suggesting a quick decay of phi29 DNA polymerase in the phage-infected bacterial lysates.

Figure 12 |RCA assay with NEB phi29 DNA polymerase or the Salmonella (109 cells/ml) lysates infected by #ST1::Ф29 at MOI=1 which were collected at 2 hr or 4 hr post infection. The fold changes were calculated by the fluorescence intensity of EvaGreen DNA binding of RCA materials without phi29 DNA polymerase as a background control. RCA was performed at 30°C for 1 hr.
To figure out the latent time and burst size of our Salmonella phage #ST1::Ф29 and the time course of RCA signals during phage infection in Salmonella, we conducted experiments that 105 cells/ml of Salmonella were infected by phage #ST1::Ф29 at MOI=1 and the lysates collected by an interval of 5 min until 60 min and subjected to plaque assays and RCA test. As Fig. 13 demonstrated, the phages were released at around 40 min (latent time) to a plateau level with a burst size of average 98.4 ± 14. Interestingly, the RCA signal increased dramatically at 35 min, achieved a high level around 40-45 min, and dropped significantly thereafter, that are consistent with our speculation of the correlation between phage lysis time (latent time) and phi29 DNA polymerase protein functionality.

Figure 13 |Salmonella phage #ST1::Ф29 latent time (min) and burst size (PFU per infected cell, the left Y axis) at MOI=1, and the relationship to RCA assay (fold change, the right Y axis). Phage-infected Salmonella lysates were harvested for 1 hour at an interval of 5 min. The lysates were subjected to plaque assays and RCA reaction followed by stained with EvaGreen dye. The burst sizes were counted by numbers of plaques. The RCA signal were read at Ex/Em=500/530 nm and divided by the background level without phi29 DNA polymerase.
To examine the feasibility in a real situation, we mimicked the drink contamination by making a serial dilution of Salmonella from 107 to 103 cells in a beaker of 500ml water and prepared the water without bacteria as a control. The water were mixed with Salmonella phage #ST1::Ф29 at the concentration of 105 PFU/500ml at room temperature for 25 min. Then, we applied our hardware of Luer lock adapter with mini Ni-column to enrich the His-phi29 DNA polymerase produced by phage-infected Salmonella. The 3D-printed Luer locker embedded a mini Ni-column was assembled onto a syringe, followed by repeatedly drawing up and pushing back the water containing phage-infected bacterial lysates in order to pass through the Ni-column. Then, the RCA materials were drawn onto the Ni-column. If His-phi29 DNA polymerase is present, the RCA reaction may be turned on. After 30 min incubation for RCA reaction, the mixtures were push back into a well of a 96-well black plate containing EvaGreen Dye in a total volume of 50 μl. The fluorescence signals were measured at Ex/Em=500/530 nm. Significant RCA signals began to appear in 2x102 bacterial cells/ml (Fig. 14). 20 cells/ml can be detected with a slight enhanced signal that is able to be distinguished from the background. However, we can’t measure the cell density under 10 cells/ml of a liquid to be examined. We think it is our limit of detection using our Salmonella phage reporter.

Figure 14 |Salmonella test at various concentrations between 2-2x104 cells/ml in 500ml water with engineered Salmonella phage carrying His-phi29 DNA polymerase gene at the concentration of 200 PFU/ml. RCA was performed on the embedded Ni-column in a 3D-printerd Luer lock adapter. The amplified DNAs were stained with EvaGreen Dye and measured at Ex/Em=500/530 nm in a microplate reader (BioTek Synergy H1).
With lots of efforts, we have successfully engineered an isolated Salmonella genome with unknown full genome sequence through Tol2 transposon system using our composite part of ldhp-Phi29 DNA pol-Tr/pTol2. The engineered phage can become a powerful Salmonella detector with highly efficient RCA reaction.
To extend the usage of BioBrick-compatible pBR322-based vector (pSB6A1) we changed the promoter of lac to ldhp and an antibiotic of ampicillin to kanamycin or chloramphenicol to generate two plasmid backbones of pSB6K1 (BBa_K3728009) and pSB6C1 (BBa_K3728010) and a composite part of ldhp-RFP-Tr/pSB6C1 (BBa_K3728013).
In our project, we focus on tackling an issue of food supply and security. Salmonella spp. are pathogens common in foodborne disease outbreaks. Salmonella typhimurium strain LT2 and the pBR322 vector with AmpR are widely used in Salmonella transgenesis studies in the laboratories9. Unexpectedly, the wild type of Salmonella labelled LT2 obtained from the lab of Prof. Cheng-Yang Huang at Chung Shan Medical University survived and grew on LB ampicillin agar plates (now we called it strain CYH). In addition, because of emergence of multidrug-resistant Salmonella10, we are motivated to develop tools for Salmonella spp. including a broad-host-range promoter and antibiotic resistance cassettes based on improving the BioBrick existing part of pBR322-based pSB6A1 (AmpR).
According to the results of antibiotics screening, we built up pSB6K1(BBa_K3728009) and pSB6C1 (BBa_K3728010), where AmpR coding region was replaced by KanR and CmR, respectively (Fig. 15). The plasmid backbones with the intrinsic BBa_J04450 standard parts between Prefix and Suffix were confirmed by growing the transformed E. coli on corresponding LB agar plates and by colony PCR check with primers on Subpart of E1010 (mRFP) and KanR gene or CmR gene (Fig. 16).

Figure 15 |Schematic plasmid maps of BioBrick pBR322-based vectors. pSB6A1 is an existing part. pSB6K1 (BBa_K3728009) and pSB6C1 (BBa_K3728010) are our new parts.
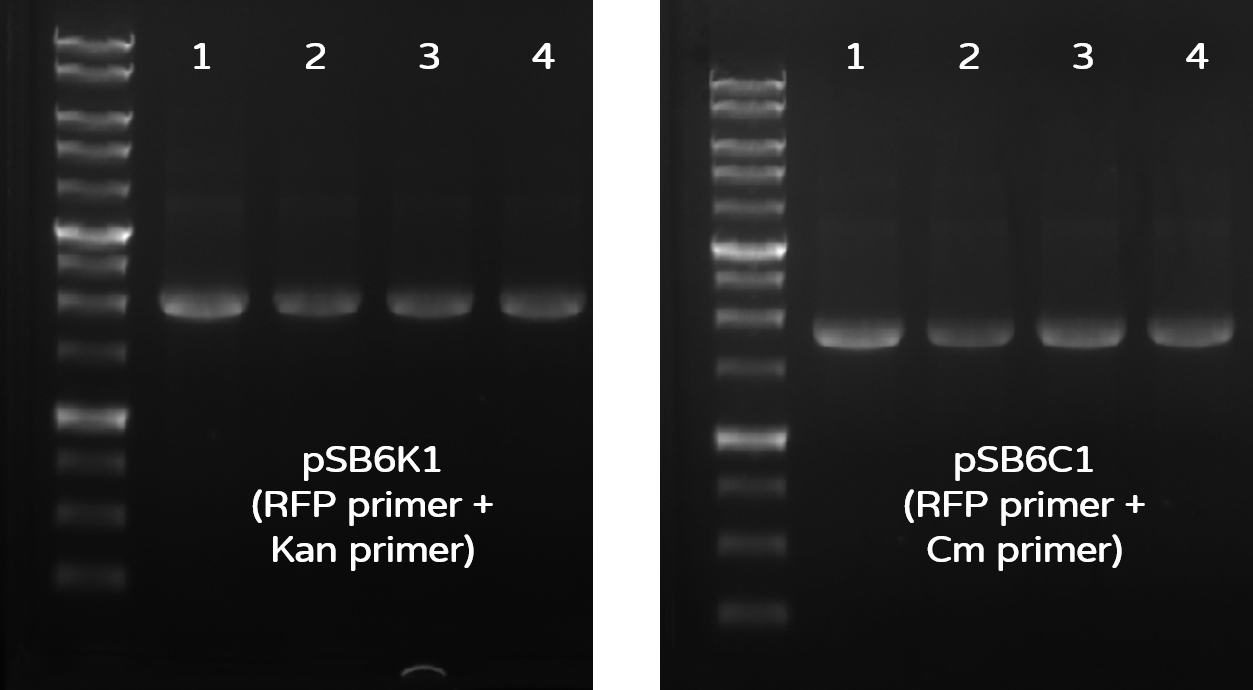
Figure 16 |Colony PCR check results on gel electrophoresis with 1 kb marker. Left gel: PCR check of 4 colonies for pSB6K1 with RFP-R + KanR-R to generate 1950-bp bands. Right gel: PCR check of 4 colonies for pSB6C1 with RFP-R + CmR-R to generate 1806-bp bands.
ldhp ( BBa_K3376000) is a strong and constitutive promoter created by us in iGEM 2020. Its activities were characterized in in vitro transcription-translation (TXTL) assay and in E. coli, Salmonella spp. and Gram-positive bacteria such as S. mutans. Moreover, pBR322-based vector is widely used to create shuttle vectors between Gram negative and positive bacteria11. Therefore, we further extended the usage of the vector by addition of the broad-host-range promoter of ldhp to replace the lac promoter. The resulting ldhp-RFP-Tr/pSB6C1 (BBa_K3376013) was confirmed by restriction enzyme check (Fig. 17).

Figure 17 |Restriction enzyme check of ldhp-RFP-Tr/pSB6C1 with EcoRI and PstI. The plasmids were prepared from 3 colonies of overnight culture of the transformed E. coli. Gel showed the 3817-bp and 1092-bp bands by restriction enzyme cuts.
Finally, the ldhp-RFP-Tr/pSB6C1 is able to transform Salmonella by electroporation at a single pulse of 12.5 kV/cm (2.5kV, 200Ω, 25μF) 9 to become chloramphenicol resistant and red colonies on a LB agar plate (Fig.18), whereas the existing part of pSB6A1 can not be used in an ampicillin-resistant Salmonella strain.

Figure 18 |Salmonella was transformed with the vectors of pSB6C1 (CmR) or pSB6A1 (AmpR) as a control. The result showed red colonies formed on a Cm LB agar plate with ldhp-RFP-Tr/pSB6C1 and no colonies with pSB6A1 vector.
We’ve made and characterized the BioBrick parts in detail. We hope the Engineering Toolkit for phage and Salmonella engineering can benefit iGEM projects in the future.
1. Urasaki A, Morvan G, Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics. 2006 Oct;174(2):639-49. doi: 10.1534/genetics.106.060244.
2. Kawakami K. Tol2: a versatile gene transfer vector in vertebrates. Genome Biol. 2007;8 Suppl 1(Suppl 1):S7. doi: 10.1186/gb-2007-8-s1-s7.
3. Ni J, Wangensteen KJ, Nelsen D, Balciunas D, Skuster KJ, Urban MD, Ekker SC. Active recombinant Tol2 transposase for gene transfer and gene discovery applications. Mob DNA. 2016 Mar 31;7:6. doi: 10.1186/s13100-016-0062-z
4. Tol2 transposase sequence (Oryzias latipes) at UniProt: UniProtKB - Q9PVN3 (Q9PVN3_ORYLA)
5. Shin J, Jardine P, Noireaux V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth Biol. 2012 Sep 21;1(9):408-13. doi: 10.1021/sb300049p.
6. Johne R, Müller H, Rector A, van Ranst M, Stevens H. Rolling-circle amplification of viral DNA genomes using phi29 polymerase. Trends Microbiol. 2009 May;17(5):205-11. doi: 10.1016/j.tim.2009.02.004.
7. Yue S, Li Y, Qiao Z, Song W, Bi S. Rolling Circle Replication for Biosensing, Bioimaging, and Biomedicine. Trends Biotechnol. 2021 Mar 11:S0167-7799(21)00039-1. doi: 10.1016/j.tibtech.2021.02.007.
8. Khattak S, Murawala P, Andreas H, Kappert V, Schuez M, Sandoval-Guzmán T, Crawford K, Tanaka EM. Optimized axolotl (Ambystoma mexicanum) husbandry, breeding, metamorphosis, transgenesis and tamoxifen-mediated recombination. Nat Protoc. 2014 Mar;9(3):529-40. doi: 0.1038/nprot.2014.040.
9. O'Callaghan D, Charbit A. High efficiency transformation of Salmonella typhimurium and Salmonella typhi by electroporation. Mol Gen Genet. 1990 Aug;223(1):156-8. doi: 10.1007/BF00315809.
10. V T Nair D, Venkitanarayanan K, Kollanoor Johny A. Antibiotic-Resistant Salmonella in the Food Supply and the Potential Role of Antibiotic Alternatives for Control. Foods. 2018 Oct 11;7(10):167. doi: 10.3390/foods7100167.
11. P. Trieu-Cuot, C. Carlier, P. Martin, P. Courvalin, Plasmid transfer by conjugation from Escherichia coli to Gram-positive bacteria, FEMS Microbiology Letters. 1987 Dec;48(1-2):289-294. doi: 10.1111/j.1574-6968.1987.tb02558.x.