Team:SCUT-China/Software

Loading...


Background
Most eukaryotic genomic DNA is wrapped in nucleosomes, which occlude and strongly distort the wrapped DNA. Transcription factor is a special protein that binds to a specific region of the gene to start transcription downstream. However, in the chromosomal state, the genomic DNA is tightly bound to the nucleosome. It can not reveal the transcribed factor binding sites (TFBS), the upstream activating sequence (UAS) for the promoter. Only when the nucleosome is relaxed or not tightly bound could TF have more opportunity to bind UAS and function effectively.
The affinity of nucleosomes to genomic DNA is determined by the arrangement and combination of sequence bases, so the affinity will be different if the sequence is slightly altered. Therefore, we hope to decrease the histone binding affinity score by introducing mutations to the sequence bases except for UAS and the core region. For this reason, a tool that can calculate the nucleosome affinity as an evaluation function to score the effect of the introduction of base fine-tuning is in urgent need.
Suppose there is a scoring tool that could calculate histone binding affinity theoretically. In that case, we could calculate nucleosome affinity for all the possible arrangements of bases in a fixed length. The promoter sequence in theory with the lowest nucleosome affinity sequence and highest strength could be selected. Of course, this approach has extremely high time complexity and low specificity; that's the reason we choose to combine genetic algorithm to optimize the promoter sequence iteratively.
Fundamental
The hidden Markov model (HMM) has been known for decades[1].
Given the characteristic of DNA sequence, and the accumulated pieces of evidence that DNA sequence itself can highly predict the localization of nucleosome in vivo, also the function of chromatin is closely related to the localization of nucleosome, scientists proposed using a duration Hidden Markov Model (dHMM) to predict the occupation of the corresponding nucleosomes in chromosomal DNA: divided into the nucleosome (N) state and the linker (L) state, where the length of the nucleosome state is fixed at 147 bp and the linker state is variable[2]. It is assumed that the two states must be alternately connected. A complete chromatin sequence must begin and end in the linker state, training a 4th order time-dependent Markov chain for the N state and a homogeneous 4th order Markov chain for the L state.
In the detailed method, they let e = e1, ..., e147 be corresponding to the nucleosome DNA sequence, and let PN be the probability of observing e as a nucleosome, computed as the product of probabilities for both Watson and Crick strands under the 4th order Markov Chain model. They assume that the linker DNA length of a given species has an unknown distribution FL (k) defined for k = 1, ..., τL (the maximum linker length allowed). An observed linker DNA sequence e = e1, ..., ek carries two pieces of information, the length is k bp, and given which, the emitted letters are e1, ..., ek . Let GL (e|k) denote the homogeneous Markov chain model for the linker DNA (again including both strands). Then observing e as a linker DNA has probability
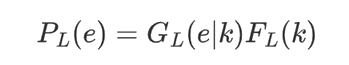
Suppose x = x1, ..., xn is a genomic DNA sequence of length n, where xi = A/C/G/T. Let z = z1, ..., zn be the corresponding hidden state path, where zi = 1 if xi is covered by a nucleosome state, and 0 otherwise. Suppose that the path z = z1, ..., zn partitions x into k consecutive nucleosome or linker state blocks, in which the nucleosome blocks have a uniform length of 147 bp, whereas the length of linker blocks may vary. They denote these blocks as y = y+, ..., yB , and their state identification as s = s1, ..., sB , where si = 1 if yi is nucleosome state, and 0 otherwise. The probability of observing (x, z) is given by
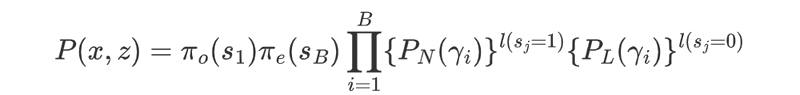
where π0(s1) and πe(sB ) stand for the probabilities that the chain initializes and ends with state s1 and sk respectively, and I is an indicator function. Since they assume that a complete chromatin sequence must start with and end in a linker state, π0(s1 = 0) = πe (sB = 0) = 1. Define the nucleosome occupancy at a specific position i, denoted o i , as the posterior probability that zi = 1, i.e.,
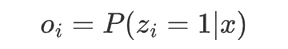
Most importantly, the histone binding affinity score at position i is defined as the log likelihood ratio for the region xi-73, ... xi , ..., xi+73 to be a nucleosome vs. a linker, i.e.,
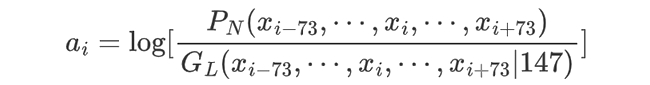
Given the models PN, GL and FL, the optimal path z can be found by the standard Viterbi algorithm, and the nucleosome occupancy score can be estimated using forward and backward algorithms[2].
The model has been trained on a large number of yeast nucleosome DNA fragments dataset that have been pyrosequencing and on a species by species basis, and the test results are satisfactory[2]. They made the nucleosome occupancy predicting tool into a software tool called NuPoP, which means "nucleosome position predicting", as an R language package for researchers to use.
Strategy
Optimizing design
We designed to obtain the variants with different nucleosome affinity of the promoter sequence by introducing the limited amount of random bases mutation without destroying the UAS sequence and the core region of the promoter.
Actually, a similar iterative optimization model has been proposed, called NucOpt Progs. The theory is technically known as a “Greedy algorithm”, in which all possible random single mutations are enumerated on the initial promoter sequence, and every single sequence variant was calculated the nucleosome affinity by NuPoP to obtain a score. After that, the sequence with the lowest affinity was selected as the winner in this round, then taken as the parent placing into the next round, calculating all mutation possible cases enumeration on this winner and vote in a new champion variant candidate. Finally, the theoretical best optimal sequence which is difficult to gain higher score is obtained[3].
However, after learning and testing NucOpt Progs, we found that this method has many disadvantages. First, the exhaustive enumeration method brings immense time complexity. It takes 36 hours to optimize a sequence of 800 bp length (based on the computer capacity). Secondly, single mutation only and greedy algorithm in use obviously determine that it's extraordinarily easy to get optimization fallen into a local optimal solution. For example, assuming that a single base change is a component of a group of mutations that significantly decrease histone affinity, but rarely calculating the benefit exactly this single base change could bring is unremarkable. In this case, the superior choice would easily be neglected. Thirdly, taking into account the biological effect, other published studies have shown that rarely low affinity is not directly related to the promoter's strength[3]. At the same time, the researchers speculate, the site distribution of nucleosome affinity may play a vital role in the promoter strength.
Given the above three defects, we have designed a software NuPGO which can make up for these shortcomings and achieve a better optimization effect. Our new strategy is to combine NuPoP with the genetic algorithm(Fig.1) using the roulette wheel operator so that the base number of mutations can realize adaptive convergence according to probability instead of only introducing a single random mutation at a time, which not only effectively reduce the time complexity but also take much more possibilities into account, escaping local optimal solution. Considering the biological effect, we additionally set the user-defined region weights especially, which enable the algorithm to place larger or smaller weights on the specific sequence region.
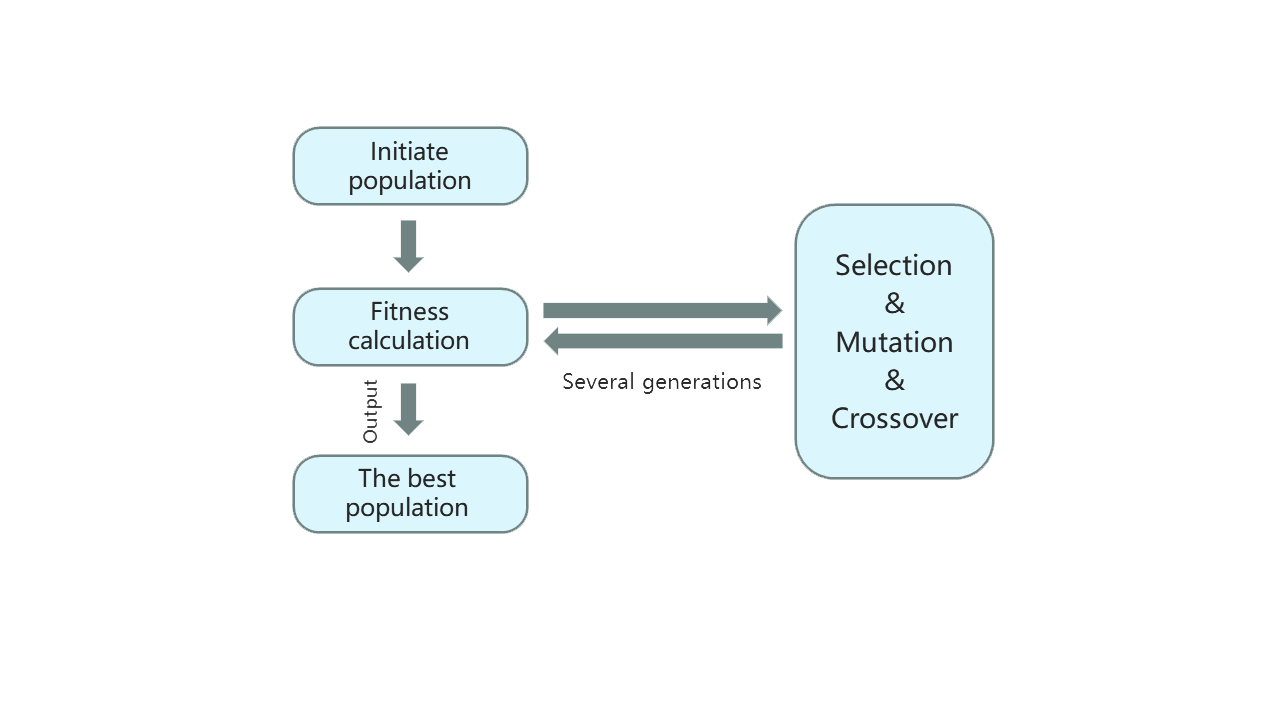
Figure 1. Genetic algorithm schematic diagram
Greedy Algorithm vs Roulette Operator
Greedy algorithm and roulette wheel selection algorithm are both essentially evolutionary algorithms. The greedy algorithm also performs better than other evolutionary algorithms in some cases. Still, its efficiency advantage brought by enumeration couldn't display here, while the multi-factor considering ability in wheel selection algorithm stands out.
Greedy Algorithm
The fatal disadvantage of the previous promoter optimizing algorithm is that only the best performer is selected in each round. Perhaps the author aimed to reduce the time complexity of exhaustive enumeration and simply pursue the best score. But such algorithms can easily trap the results into local optima. For example, the three-point mutation on site ABC decrease the histone affinity more than the three-point mutation on site DEF. However, the affinity decrease of site B mutation is smaller than that of site D mutation, so the greedy algorithm will choose site D mutation strategy as the winner, while site B strategy is obviously better actually.
Moreover, only a random single mutation is introduced in each round. In other words, the mutation number is set to 1, which leads to gigantic amounts of enumerations, and the time complexity has not been sufficiently reduced. The execution of the greedy algorithm is time-consuming. In our test, an 800bp length promoter had to cost 36 hours to run.

Figure 2. Schematic diagram of NucOpt Progs
Roulette Operator
The wheel selection method is also known as the fitness proportionate selection (FPS), which means that the fitness of an individual is directly proportional to the probability of being selected, similar to putting all the individuals on a wheel, the area occupied by each individual is directly proportional to its fitness, that is, the probability of being selected is determined by the area of the plate, namely the fittness of each individual when the wheel spins randomly.
In other words, the fitness is directly proportional to the probability of being selected, turning the wheel N times to select N individuals. Meanwhile, it also means that an individual can be selected repeatedly.

Figure 3. Schematic diagram of genetic algorithm applied to promoter optimizer NuPGO
For random bases mutations introduced in each round, we set the mutation amount as an adaptive parameter. Each base position in the sequence is a probability mutation rather than the unintelligent setting the amount as "1" from beginning to end. To ensure the rationality and accuracy of the optimization, we set the threshold of mutation probability to 0.0005 so that the number of base mutations in each round would not exceed 5. Of course, other choices are worth expanding. By doing this, the time complexity of the model is reduced to some degree, the curve will converge faster when performing the gradient descends, broadening the search, and more combinations would be explored to approach the global optimal solution. As the iteration converges, the mutation probability will gradually approach 0, that is, reaching the final optimized promoter sequence(Fig.3).
Specific Weights
The critical UAS are usually only about 10 bp in length, and the vital goal we want to achieve, precisely speaking, is to reduce the nucleosome affinity around the UAS region and to make the UAS region tend to become the linker region rather than nucleosome region.
The final goal of the algorithm is to decrease the total affinity score of the whole promoter sequence as far as possible, which is likely to ignore the key UAS region mutation and choose other strategies with more significant benefit, the final optimized promoter obtained in such condition may not make use of the advantages of UAS. Specifically, the key UAS is still in the high histone affinity region. To compensate for this shortcoming, we decided to artificially add a regional weight to the score calculation process. According to the formula introduced above, dHMM calculates the nucleosome affinity score for every single site, and then calculates the area under the curve (AUC).
To make some adjustments, we decide to compute the score of every single position with a custom zoom when calculating the specific area that we regard special, such as the area of the crucial UAS, in other words, multiplying the simply calculated score by the user-defined weight to get each final single point score.
Fortunately, the results of testing different weights on the NuPGO algorithm are satisfactory. After adding weights to a specific area, NuPGO will pay more attention to that area. In the final optimized result sequence, this specific region will get better performance of nucleosome affinity degradation.
Sustainable
Ultimately, and most worth noting is that, neither the NuPoP software program which can evaluate DNA’s nucleosome affinity[2] nor the iterative optimization code[3] that uses the greedy algorithm, because of the components missing and internet limitation, following the tutorials provided in the papers, we couldn’t get the complete executable code file and failed to run the optimizing software with greedy algorithm directly, as well as other researchers. It means the use of NuPoP and greedy algorithm optimizer have been difficult for those who lack the computer or bioinformatics study infrastructure.
Therefore, we refactored the implementation code almost completely according to some of its logic, and wrote the genetic algorithm using the roulette wheel operator, which is much easier to read and user-friendly much more than the previous code. In other words, we fix the algorithm published before and come up with a more novel and intelligent software. Both the greedy algorithm optimizer NucOpt Progs and genetic algorithm optimizer NuPGO are published by us for users’ convenient.
This means that anyone, especially iGEM teams in the future and researchers in labs, can easily get started using our program based on a tutorial. Input your initial data and conditions visually and easily and it will be on. Those interested are also welcomed to read our source code and make some comments or modifications, and maybe a stronger NuPGO will be come up with next year.
Result Analysis
Optimization result on our promoter
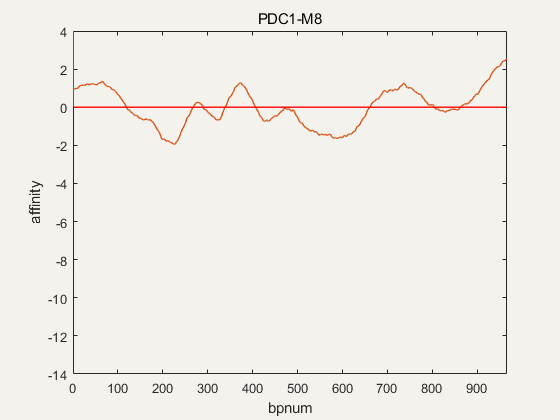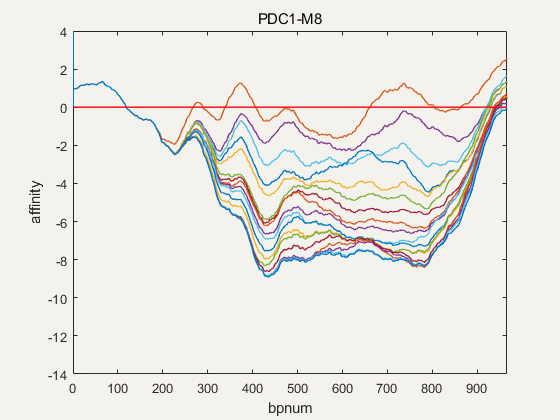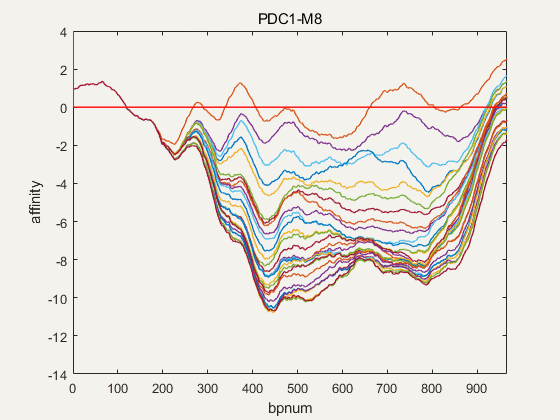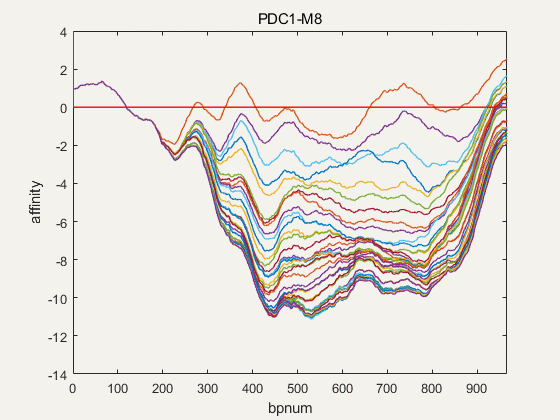
Figure 4. NuPGO optimization on our project promoter nucleosome affinity
Time-consuming comparison

Optimization effect comparison
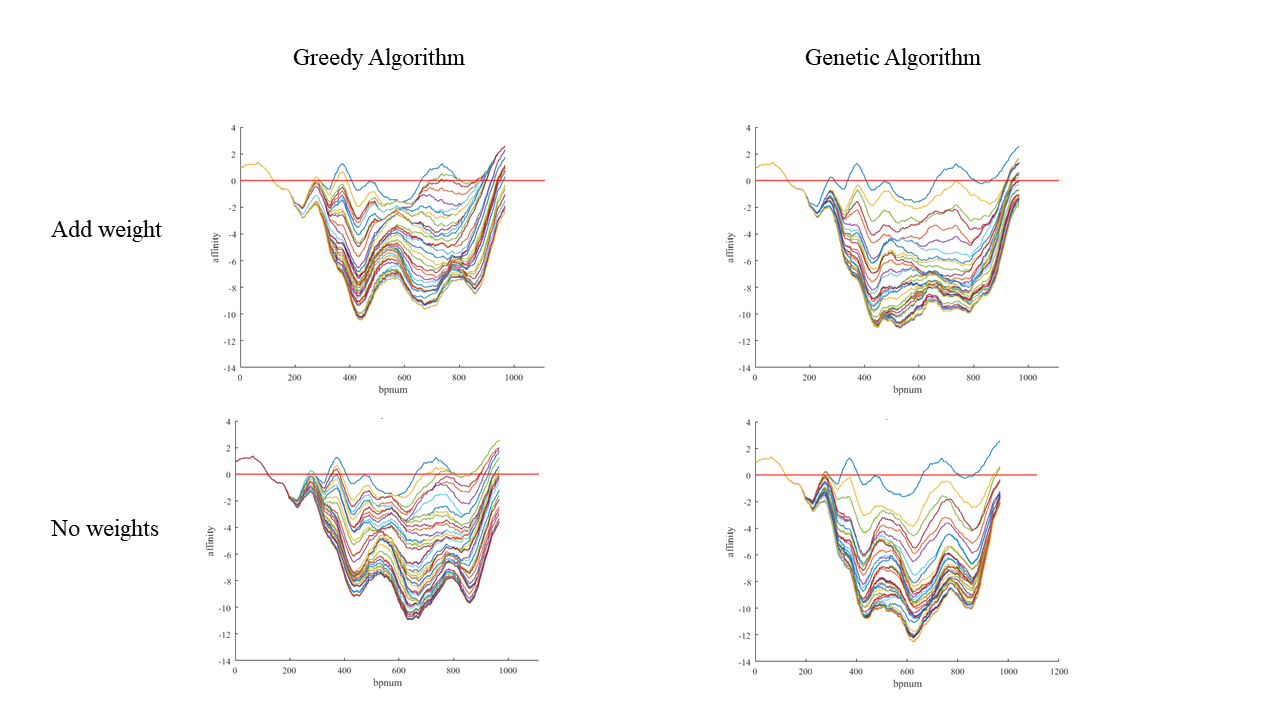
Figure 5. Result comparison between Greedy Algorithm and Genetic Algorithm, weights added result and result without weights.
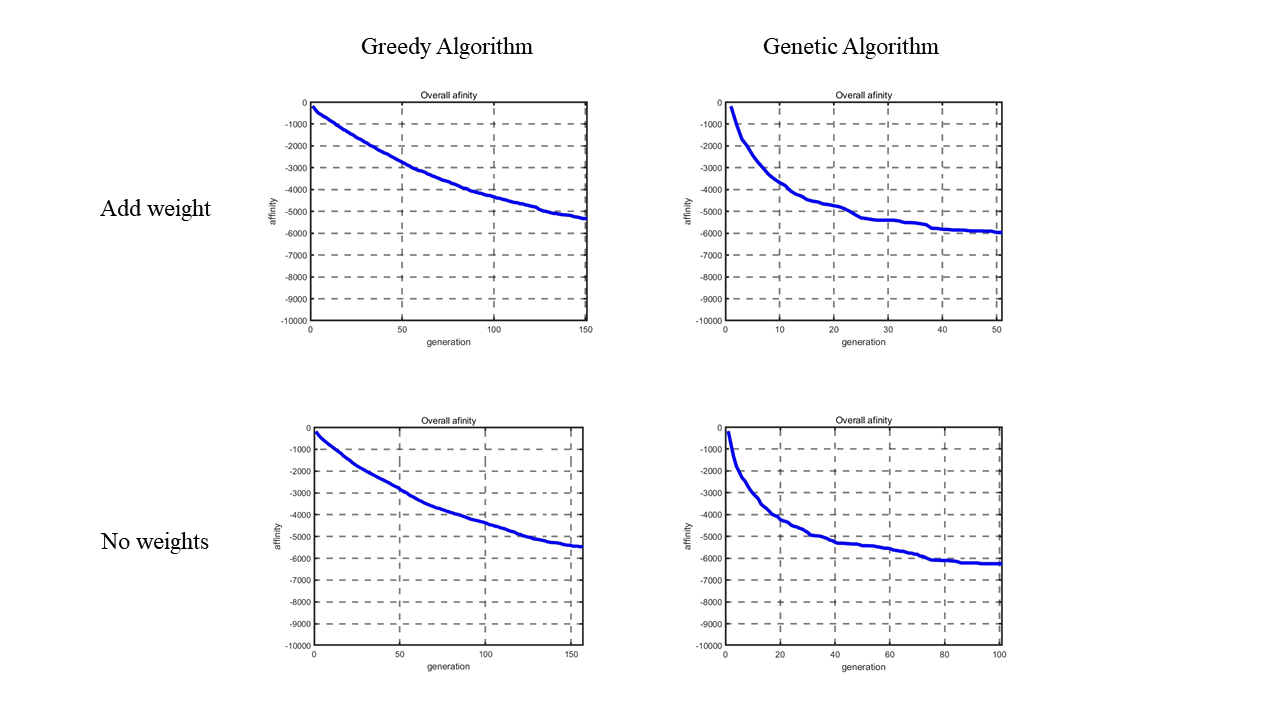
Figure 6. Descent curve of total nucleosome affinity score between Greedy Algorithm and Genetic Algorithm, weights added result and result without weights.
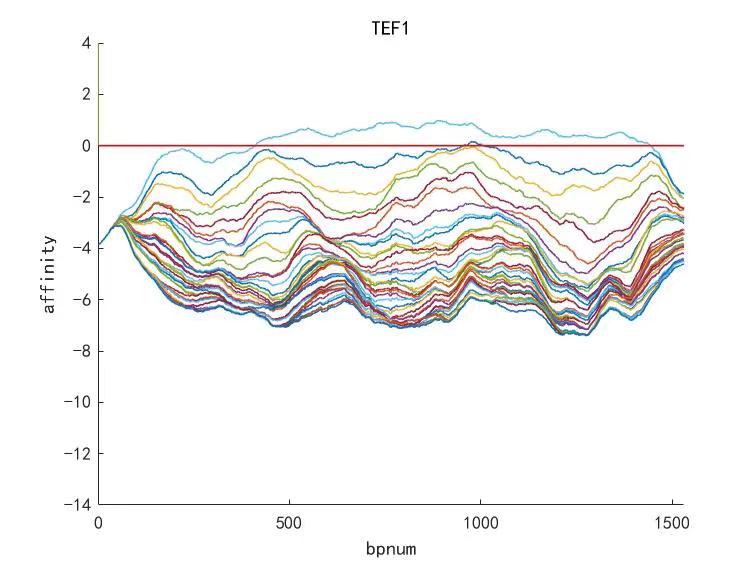
Figure 7. Other example of promoter TEF1 optimizing
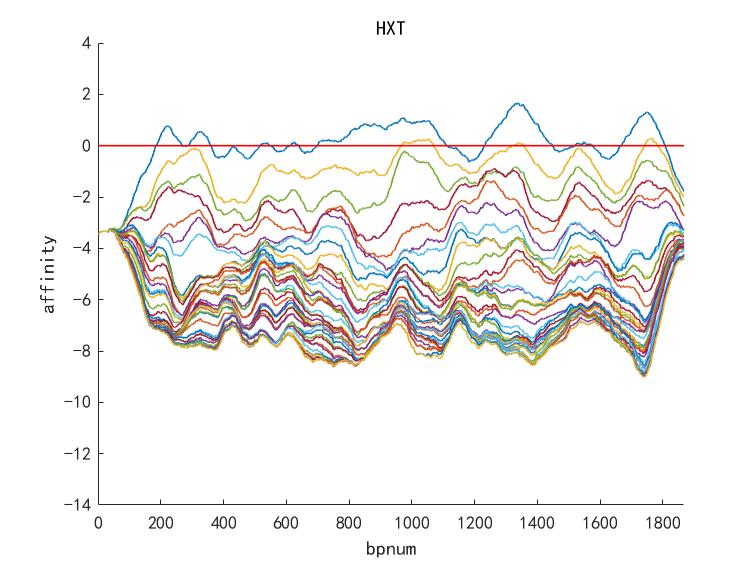
Figure 8. Other example of promoter HXT1 optimizing
NuPGO result visualization

Figure 9. Comparison between initial and optimized project promoter M8 sequence
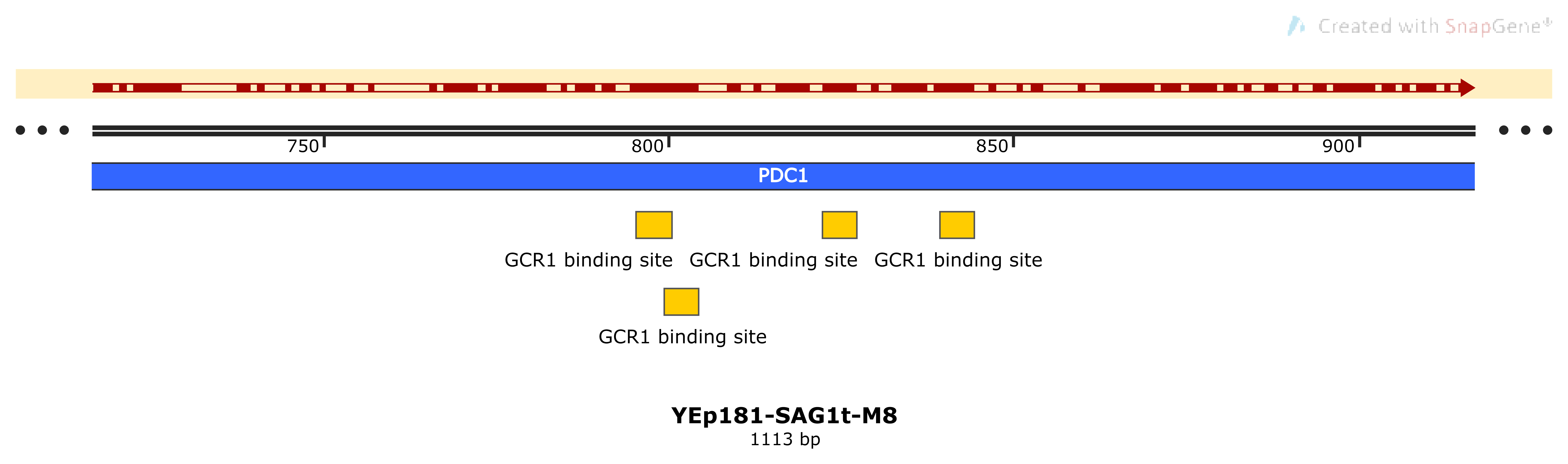
Figure 10. Partial protected UAS region during optimization
NuPGO result verification
Unfortunately, the NuPGO-optimized promoter has some mutation of around hundreds of bp, requiring chemical synthesis to obtain the new promoter gene fragment. Moreover, it is not statistically representative of testing only the final variant. To prove that the strength of the NuPGO optimized promoter sequence is consistent with the predicted results, we need to select at least 5 to 10 promoter variants, and conduct chemical synthesis and characterization experiments to verify their strength. Due to the lack of project funds and time constraints, we can not perform characterization experiments to ascertain the strength of the sequence obtained by the optimization algorithm.
If conditions permit or any other team was interested in verifying the effect of NuPGO, we would like to or recommend selecting the promoter variants with 50, 100, 200, 400, 500bp mutations in the same run, also pick three variants at each of the position, such as 499, 500, 501bp. Then design the parallel characterization experiments to see the strength difference. The descent curve could also be consulted to obtain the selection choice strategy.
Nevertheless, we have reason to believe that the results of the optimization are relatively reliable, because, in our references(Fig.6), the authors conducted experiments on the strength characterization of the promoter variants obtained by the greedy algorithm. We can conclude from the experimental results that nucleosome affinity optimization indeed can increase the promoter's strength and thus enhance expression.
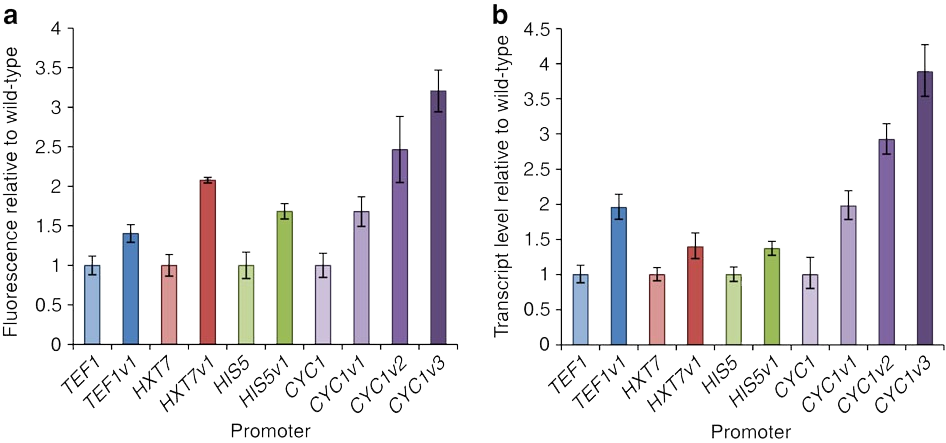
Figure 11. Redesign of native yeast promoters for increased expression by decreasing nucleosome affinity in reference article. DOI: 10.1038/ncomms5002
Availability
We publish our source code and packages on Github. We extraordinarily sincerely and strongly recommend that every future iGEM team or researcher using a eukaryotic promoter use our software to achieve a higher expressing level and rational use of UAS strong promoters.
GithubReferences
[1]L. R. Rabiner, "A tutorial on hidden Markov models and selected applications in speech recognition," in Proceedings of the IEEE, vol. 77, no. 2, pp. 257-286, Feb. 1989, doi: 10.1109/5.18626.
[2]Xi, L., Fondufe-Mittendorf, Y., Xia, L. et al. Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinformatics 11, 346 (2010). https://doi.org/10.1186/1471-2105-11-346
[3] [3]Curran, K., Crook, N., Karim, A. et al. Design of synthetic yeast promoters via tuning of nucleosome architecture. Nat Commun 5, 4002 (2014). https://doi.org/10.1038/ncomms5002
[4]de Boer, C.G., Vaishnav, E.D., Sadeh, R. et al. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat Biotechnol 38, 56–65 (2020). https://doi.org/10.1038/s41587-019-0315-8.