Team:Evry Paris-Saclay/Engineering
Engineering Success
Experiments & Results
Engineering
Evolution.T7 system is an improved directed evolution strategy based on the use of T7RNAP. The system has the goal of making improvements over the properties and performance of previous directed evolution methods based on T7RNAP, such as MutaT7 [1], T7-DIVA [2], and eMutaT7 [3]. More precisely, Evolution.T7 took inspiration from the T7-DIVA tool, where a T7 promoter placed downstream of the gene of interest allows the T7RNAP fused with a base deaminase to generate random mutagenesis until reaching a dCas9 roadblock [2].
In addition, our strategy follows the search for an increased mutation types and rates, as the approach followed by the newly published TRIDENT [4] system, which even achieved T to G transversions. The systems mentioned above have several biases as mutations are mainly limited to the non-template strand and transitions are the main types of mutations. Therefore, when setting out to build the Evolution.T7 system, we took these biases and limitations into consideration to design an improved directed evolution system.
The Evolution.T7 project was inspired and based on the engineering framework of Synthetic Biology: Design, Build, Test, and Learn (The DBTL cycle). It is possible to fit the project in the DBTL cycle, which helped the team progress, including data analysis of the system.
Evolution.T7 DBTL Cycle
Design
The design behind the Evolution.T7 tool aimed to overcome the two previously mentioned biases. For this purpose, the mutagenic system also implemented two specific considerations (i) effecting mutations on both strands, (ii) higher mutation rate, and (iii) increased diversity in the type of mutation. Briefly, the mutators used are adenine or cytosine deaminases creating intermediates that lead to transition mutations (A -> G or C -> T and vice versa). The process is illustrated in Figure 1.
Since the previous methods mainly made transitions, and only in one of the DNA strands, the aim of generating mutations in both strands was to increase the rate of those mutations. More precisely, the first improvement of the system considered flanking the region to evolve with two different promoters recognized by different T7RNAPs (Figure 2). This design enables the induction of the two T7RNAPs by two different inducers, allowing mutations in both strands while avoiding collision of mutagenesis complexes by temporally separating their induction. As the second orthogonal T7RNAP, we chose to use the T7RNAP-CGG-R12-KIRV [5] which is a mutant T7 RNA polymerase version that transcribes from the mutant T7CGG promoter, while the wt T7RNAP is the natural polymerase that transcribes from the T7wt promoter.
The system generated many versions of T7RNAP fusions to previously used base deaminases (AID, pmCDA1, rAPOBEC1, TadA*) and enhanced variants (ABE8.20m [6], evoAPOBEC1-BE4max [7], evoCDA1-BE4max [7]) to increase the mutation diversity and rate.

Figure 1. Exemplification of the adenine or cytosine deamination as a source of mutation in one of the phenylalanine codon TTT (A) and one arginine codon CGT (B).
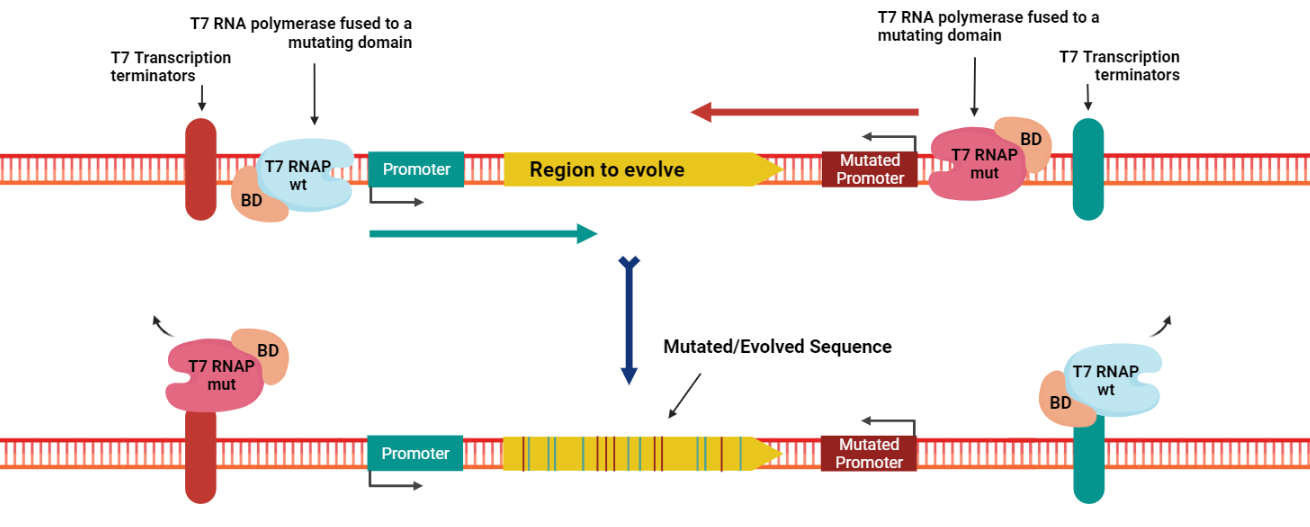
Figure 2. The principle of Evolution.T7 as a tool. Image created in BioRender.com.
The system followed a combinational approach for the mutators searching to increase the mutation rate and diversity. In detail, the combination included eight possible fusions for each of the two T7 RNAP, having a total of 64 combinations tested. To evaluate all the possibilities and further select the best combinations, we developed a deterministic model based on the genetic diversity as a function of time (see the "Modeling" section of this wiki for a detailed description).
Finally, to test the Evolution.T7 system, we created a testing or reporter plasmid having a dual strategy to evaluate the mutagenic capacity of our tool. This plasmid has two fluorescent proteins next to each other in opposite directions (sfGFP in the forward and mCherry in reverse direction) and an ampicillin resistance gene, as shown in Figure 3. This genetic circuit is flanked by the two T7 promoters delimiting the zone to be mutated: the T7wt promoter in forward orientation and the T7CGG promoter in reverse orientation. In addition, the T7 promoters are flanked by four different strong T7 terminators to ensure the T7RNAP stops and avoids mutations in the rest of the plasmid (four terminators have been shown to be required for efficient transcription termination of T7RNAP [1]).

Figure 3. Evolution.T7 testing plasmid design for a dual selection system (BBa_K3766111 assembled in pSEVA73 backbone). Image created in BioRender.com.
Build
The strategy followed by Evolution.T7 required two plasmids carrying the two different versions of the T7RNAP, with or without fusion with different mutators, and the reporter or testing plasmid mentioned before, having the target gene to evolve. First of all, the team from Dr. Luis Ángel Fernández, at Centro Nacional de Biotecnología (CNB-CSIC) in Spain, kindly provided us with 5 plasmids constructed for the T7-DIVA tool [2]. These 5 plasmids encode the T7RNAP fused with 4 base diversifying domains (AID, pmCDA1, rAPOBEC1, TadA*), and 1 unfused T7RNAP control, under the control of the pTetA promoter in the pSEVA22 backbone [8].
Nevertheless, as the Evolution.T7 requires two different T7RNAP, we assembled a mutated T7RNAP (T7RNAP-CGG-R12-KIRV) fused with the different mutator domains under the control of the pBad promoter in the pSEVA73 backbone [8]. In addition, the enhanced mutator variants (ABE8.20m, evoAPOBEC1-BE4max, evoCDA1-BE4max) were fused with the wild-type T7RNAP (under the control of the pTetA promoter in the pSEVA22 backbone [8]) to complete the part collection of the system. All Evolution.T7 plasmids are listed in Table 1. They were assembled by Golden Gate using BsaI or BsmBI restriction enzymes from PCR fragments amplified from the 5 plasmids provided by CNB-CSIC and gBlocks synthesized by IDT.
Table 1. Evolution.T7 plasmids used and assembled by Golden Gate.
| Label | Fusion Protein Basic Part Numbers | Composite Part Number or Accession | Promoter | Plasmid backbone |
|---|---|---|---|---|
| T7RNAP | - | GenBank MN450171 | pTetA | pSEVA22 |
| AID-T7RNAP | - | GenBank MN450172 | pTetA | pSEVA22 |
| pmCDA1-T7RNAP | - | GenBank MN450173 | pTetA | pSEVA22 |
| rAPOBEC1-T7RNAP | - | GenBank MN450174 | pTetA | pSEVA22 |
| TadA*-T7RNAP | - | GenBank MN450175 | pTetA | pSEVA22 |
| evoAPOBEC1-BE4max-T7RNAP | BBa_K3766008 | BBa_K3766108 | pTetA | pSEVA22 |
| evoCDA1-BE4max-T7RNAP | BBa_K3766009 | BBa_K3766109 | pTetA | pSEVA22 |
| ABE8.20-m-T7RNAP | BBa_K3766010 | BBa_K3766110 | pTetA | pSEVA22 |
| T7RNAP-CGG-R12-KIRV | BBa_K3766000 |
BBa_K3766100 | pBad | pSEVA47 |
| AID-T7RNAP-CGG-R12-KIRV | BBa_K3766001 | BBa_K3766101 | pBad | pSEVA47 |
| pmCDA1-T7RNAP-CGG-R12-KIRV | BBa_K3766002 | BBa_K3766102 | pBad | pSEVA47 |
| rAPOBEC1-T7RNAP-CGG-R12-KIRV | BBa_K3766003 | BBa_K3766103 | pBad | pSEVA47 |
| TadA*-T7RNAP-CGG-R12-KIRV | BBa_K3766004 | BBa_K3766104 | pBad | pSEVA47 |
| evoAPOBEC1-BE4max-T7RNAP-CGG-R12-KIRV | BBa_K3766005 | BBa_K3766105 | pBad | pSEVA47 |
| evoCDA1-BE4max-T7RNA-CGG-R12-KIRV | BBa_K3766006 | BBa_K3766106 | pBad | pSEVA47 |
| ABE8.20-m-T7RNAP-CGG-R12-KIRV | BBa_K3766007 | BBa_K3766107 | pBad | pSEVA47 |
Test
To evaluate the performances of our Evolution.T7 targeted directed evolution system, we decided to use E. coli MG1655 cells harboring two mutations Δung and Δnfi that were shown to be important for enhancing the mutation rate of the mutagenic system [2]. ung is a gene encoding the uracil DNA N-glycosilase that is involved in the elimination of uracil from DNA molecules [9], while nif is a gene encoding the endonuclease V of E. coli which eliminates inosines [10,11]. Uracil and inosine are nucleobases produced by the action of cytosine and adenine deaminases, respectively, which are the mutator domains of our Evolution.T7 system.
E. coli MG1655 Δung Δnfi [2] (kindly provided by Dr. Luis Ángel Fernández, at Centro Nacional de Biotecnología (CNB-CSIC) in Spain) were first transformed with our reporter plasmid (BBa_K3766111 in pSEVA73 backbone [8]), together with different combinations of forward and reverse mutators in pSEVA22 and pSEVA47 backbones, respectively (Table 1, Figure 4).

Figure 4. Schematic representation of the Evolution.T7 system. Image created in BioRender.com.
Transformed bacteria were first grown overnight in 96-deep-well plates containing 1 mL of LB medium supplemented with the relevant antibiotics (5 µg/mL trimethoprim, 12.5 µg/mL kanamycin and 12.5 µg/mL spectinomycin), then diluted by 40x into similar media. Upon reaching early log-phase, cells were further diluted 20x in LB medium supplemented with the antibiotics (5 µg/mL trimethoprim, 12.5 µg/mL kanamycin, 12.5 µg/mL spectinomycin), and inducers (200 ng/mL anhydrotetracycline and 1.5 mM L-arabinose). Starting from this point, 3 types of tests were performed:
- Fluorescence measurements. For this, the 20x dilution was performed in 100 µL in a 96-well microplate. The plate was incubated at 37 °C at 200 rpm and the sfGFP fluorescence (λexcitation 483 nm and λemission 530 nm), the mCherry fluorescence (λexcitation 570 nm and λemission 620 nm), and OD600nm were measured every 10 min for 24 hours in a CLARIOstar (BMGLabtech) plate reader. Fluorescence values were normalized by OD600nm and, using the calibration curves presented on the "Measurement" page of this wiki, we converted the arbitrary units into Molecules of Equivalent FLuorescein (MEFL) / particle or Molecules of Equivalent Texas Red (METR) / particle, respectively.
- NGS sequencing. For this the 20x dilution was performed in 10 mL, and after an overnight incubation at 37 °C and 200 rpm, plasmids were extracted using the Wizard Plus SV Minipreps DNA Purification System (Promega) and sent to Novogene Co. Ltd for further processing. Novogene Co. Ltd first performed a quality control by capillary electrophoresis on a 5400 Fragment Analyzer system (Agilent) for quantitation and assessing sample’s integrity and purity, followed by library construction, quality control and sequencing. For this, DNA was randomly sheared into short fragments, then they were end repaired, A-tailed and further ligated with Illumina adapter.
The fragments with adapters were PCR amplified, size selected, and purified. The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries will be pooled and sequenced on Illumina platforms, according to effective library concentration and data amount required. - Selection of AmpR gain of function mutants. For this, the 20x dilution was performed in 100 µL in a 96-well microplate. The plate was incubated at 37 °C at 200 rpm and 5 µL of cell suspensions were spotted, 1, 3 and 24 hours later, onto LB agar plate containing increasing concentrations of aztreonam.
Learn
Evolution.T7 modifies GFP and mCherry fluorescence signals
The first experiment performed to validate the mutagenic capacity of the Evolution.T7 tool was to analyze the fluorescence produced by the reporter plasmid during evolution. As previously described, the reporter plasmid (Figure 3) contains sfGFP and mCherry proteins in opposite directions. They are flanked by the two T7 promoters: the T7wt promoter in forward orientation to drive the expression of sfGFP by the T7RNAP and the T7CGG promoter in reverse orientation to drive the expression of mCherry by the T7RNAP-CGG-R12-KIRV. As controls, we used empty plasmids carrying none of the T7RNAP enzymes, indicated by the top left squares of each heatmap in Figure 5.
Analyzing these heatmaps, one can first observe that both the sfGFP and mCherry expressions were readily detected, although the expression is not very intense compared to the negative controls, most probably due to the particular orientation of the genes of the two fluorescent proteins and the strength of their respective RBSes. Indeed, the design of the two RBSes (BBa_K3766033 and BBa_K3766034) was challenging as they are next to each other in back to back orientation and this required multiple rounds of design using the Salis Lab / De Novo DNA RBS Library Calculator v2.1.1 [12–14]. Nevertheless, this expression proves that both polymerases are expressed and the reporter system is functional.
Moreover, we observe variations in the sfGFP and mCherry signals due to the mutations caused by the mutagenesis modules. These variations are either a decrease or an increase of the fluorescence production compared to the controls. Theoretically, mutations should happen randomly along the reporter genes, leading to significant variations between the replicates themselves. Thus, we expected to observe a decrease in fluorescence production in most of the cases, which it is.
However, we can also identify an increase of fluorescent signals in some cases which is likely due to mutations in the RBSses of the reporter genes.
In addition, we see that the joint action of both T7RNAP led to more extreme effects on both fluorescent signals, often completely silencing the reporter genes.This reveals the synergy between both mutator T7RNAPs to mutate a gene, and lead to believe that the evolving of the antibiotic resistance gene is also more efficient with both polymerases.

Figure 5. In vivo characterization of sfGFP and mCherry expression by E. coli MG1655 Δung Δnfi [2] harbouring the reporter plasmid (BBa_K3766111 assembled in pSEVA73 backbone) and different combinations of forward mutators (T7RNAP fused to different mutator domains in pSEVA22 backbone) and reverse mutators (T7RNAP-CGG-R12-KIRV fused to different mutator domains in pSEVA47 backbone). The data are the mean MEFL/particle and METR/particle of at least three measurements on independent biological replicates. Same data in bar charts representation with standard deviations is available here.
Deep insights in the mutagenic effects of Evolution.7 system
To further characterize the Evolution.T7 system, we sequenced by Next Generation Sequencing (NGS) 18 pools of cells after a diversification step (Figure 6). One for each mutator fused with one of the two polymerase (T7RNAP and T7RNAP-CGG-R12-KIRV) and the negative controls containing either only one RNA polymerase or none at all.

Figure 6. Protocol for the NGSequencing of the 18 pools after diversification.
The details of the analysis of these sequencing data is available in the "Modeling" part of this wiki. We first checked the activity of the mutators according to two criteria: the type of mutation generated and their number (greater than the pools without mutators) (Figure 7).
Taking into consideration that the deamination occurs mostly on the non template strand ([2]), the expected results for the action of different mutators are:
- cytosine deamination on forward orientation: C -> T
- cytosine deamination on reverse orientation: G -> A
- adenine deamination on forward orientation: A -> G
- adenine deamination on reverse orientation: T -> C
The specificity of the mutators is globally respected, the tricky point is that even for the adenine deaminases, the transitions A -> G have a lower occurrence. At the same time, the number of specific mutations for each mutator is significantly higher (in number and abundance) than without mutators (empty plasmid or only RNA polymerase).




Figure 7. SNPs spotted in the NGS analysis. Each point is a mutation found in the pool of the corresponding mutator
Then, we wanted to compare our new mutators and our new polymerase to the previously described tools. Helped by our "Modeling" part, we calculated an indicator of the mutation rate for each couple of polymerase and mutator (Figure 8). We can see that the T7RNAP-CGG-R12-KIRV polymerase fusions seem to induce on average more mutations. But as we do not especially want the higher mutation rate but the higher diversity inside our pool of cells, we linked this rate to an estimator of the diversity ("Modeling") in order to select the best couple of mutators. At our timescale (less than 20 generations), we found that the mutation rate is closely correlated to the diversity (see our "Modeling" page of this wiki for more details)

Figure 8. Mutation rates (mutations per kilobase and per generations) of the Evolution.T7 system with every mutator based on the analysis of Next Generation Sequencing.
From Ampicillin to Aztreonam resistance using Evolution.T7 mutagenic system
To further validate the Evolution.T7 as a directed evolution tool, we characterized the evolution of the AmpR gene, which corresponded to the second part of the reporter plasmid that was included in the target evolution region flanked by the two T7 promoters (Figure 3). All the experimental details and the sequential results obtained for this experiment are detailed in the "Proof of Concept" page on this wiki). Nevertheless, these results showed the success of the mutagenic system essential in our engineering success.
First, among the three times tested for the rounds of evolution, 24 hours gave the highest mutant colonies when grown with aztreonam as the selection method. From these colonies, the significant majority grew with 10 µg/mL of Aztreonam, and even four colonies achieved to propagate with 20 µg/mL of the antibiotic in the media (Figure 9).
Among the different mutators, the evoAPOBEC1-BE4max-T7RNAP-CGG-R12-KIRV led to the highest number of mutants, followed by pmCDA1-T7RNAP-CGG-R12-KIRV. From this experiment, a total of 35 mutants resistant to aztreonam were further analyzed. The sequence analysis revealed at least one mutation and up to 10 compared to the original AmpR sequence, leading to reported and new variants of aztreonam resistant strains (detailed analysis in "Proof of Concept" page on this wiki).
Therefore, this analysis confirmed the previous observations regarding fluorescence variations on the activity of our mutagenic tool. The Evolution.T7 tool is consequently a mutagenic system, validated by the mutations identified in the mutants resistant to aztreonam after 1h, 3h and 24 hours rounds of evolution.

Figure 9. Number of aztreonam resistant colonies isolated on 10 and 20 µg/mL aztreonam plates as a function of different combinations of mutator domaines of our Evolution.T7 system.
Conclusion
Evolution.T7 is a highly efficient and easy-to-use directed evolution tool whose capacities were probed by different means: (i) the levels of the fluorescence reached by two reporter genes encoding the sfGFP and mCherry proteins after induction of the evolution system, (ii) the deep NGS sequencing of all variants that can be generated (in the absence of any selection), and finally, as a Proof of Concept experiment (ii) the possibility to generate variants of the ampicillin-resistance gene AmpR able to confer resistance to aztreonam, a different antibiotic (Check our "Proof of Concept" page on this wiki for a more detailed description).
References
[1] Moore CL, Papa LJ, Shoulders MD. A processive protein chimera introduces mutations across defined DNA regions in vivo. Journal of the American Chemical Society (2018) 140: 11560–11564.
[2] Álvarez B, Mencía M, de Lorenzo V, Fernández LÁ. In vivo diversification of target genomic sites using processive base deaminase fusions blocked by dCas9. Nature Communications (2020) 11: 6436.
[3] Park H, Kim S. Gene-specific mutagenesis enables rapid continuous evolution of enzymes in vivo. Nucleic Acids Research (2021) 49: e32–e32.
[4] Cravens A, Jamil OK, Kong D, Sockolosky JT, Smolke CD. Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nature Communications (2021) 12: 1579.
[5] Meyer AJ, Ellefson JW, Ellington AD. Directed evolution of a panel of orthogonal T7 RNA polymerase variants for in vivo or in vitro synthetic circuitry. ACS synthetic biology (2015) 4: 1070–1076.
[6] Gaudelli NM, Lam DK, Rees HA, Solá-Esteves NM, Barrera LA, Born DA, Edwards A, Gehrke JM, Lee S-J, Liquori AJ, Murray R, Packer MS, Rinaldi C, Slaymaker IM, Yen J, Young LE, Ciaramella G. Directed evolution of adenine base editors with increased activity and therapeutic application. Nature Biotechnology (2020) 38: 892–900.
[7] Thuronyi BW, Koblan LW, Levy JM, Yeh W-H, Zheng C, Newby GA, Wilson C, Bhaumik M, Shubina-Oleinik O, Holt JR, Liu DR. Continuous evolution of base editors with expanded target compatibility and improved activity. Nature Biotechnology (2019) 37: 1070–1079.
[8] Martínez-García E, Goñi-Moreno A, Bartley B, McLaughlin J, Sánchez-Sampedro L, Pascual Del Pozo H, Prieto Hernández C, Marletta AS, De Lucrezia D, Sánchez-Fernández G, Fraile S, de Lorenzo V. SEVA 3.0: an update of the Standard European Vector Architecture for enabling portability of genetic constructs among diverse bacterial hosts. Nucleic Acids Research (2020) 48: D1164–D1170.
[9] Pearl LH. Structure and function in the uracil-DNA glycosylase superfamily. Mutation Research (2000) 460: 165–181.
[10] Guo G, Ding Y, Weiss B. nfi, the gene for endonuclease V in Escherichia coli K-12. Journal of Bacteriology (1997) 179: 310–316.
[11] Vik ES, Nawaz MS, Strøm Andersen P, Fladeby C, Bjørås M, Dalhus B, Alseth I. Endonuclease V cleaves at inosines in RNA. Nature Communications (2013) 4: 2271.
[12] Reis AC, Salis HM. An automated model test system for systematic development and improvement of gene expression models. ACS synthetic biology (2020) 9: 3145–3156.
[13] Farasat I, Kushwaha M, Collens J, Easterbrook M, Guido M, Salis HM. Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Molecular Systems Biology (2014) 10: 731.
[14] Ng CY, Farasat I, Maranas CD, Salis HM. Rational design of a synthetic Entner-Doudoroff pathway for improved and controllable NADPH regeneration. Metabolic Engineering (2015) 29: 86–96.












© 2021 • iGEM Evry Paris-Saclay