Team:Evry Paris-Saclay/Description
Description of the project
Directed Evolution of Proteins
Description
Every life form emerged by evolution. Successive random mutations followed by the selection of the fittest individuals have resulted in vastly complex and diverse living organisms, from bacteria and archaea to plants or mammals, and every species is well adapted to its environment. These variations are easily observed at microscopic and macroscopic levels for all life forms, but also at the molecular level particularly in DNA, RNA and Proteins.
Inspiration
This year, the iGEM 2021 Évry Paris-Saclay team has decided to work on developing a tool for in vivo Directed Evolution of proteins. This idea came to us early on. As we were choosing a project from a pool of various interesting brainstormed projects, an issue quickly arose: a lot of those ideas required extensive protein engineering to be really innovative, and rational protein engineering requires expert knowledge of sequence-function relationships and can be very slow and tedious.
This issue inspired us to build our project to address directed evolution as the fastest and easiest way for obtaining new protein functions. Directed evolution of proteins is an alternative method that simulates natural phenomena but guides their outcome [1]. By generating random mutations and selecting for a specific desired trait or functionality. By repeating the diversification-selection process, it is possible to rapidly attain a vast diversity of proteins, often better adapted to a task than a researcher could have designed it to be.
Directed evolution tools have been applied to improve diverse types of proteins, for instance, enzymes used in industrial biocatalysis processes, or transcription factors to build biosensors able to recognize new compounds [2–4].

Existing directed evolution tools and their limitations
These methods can be categorized into in vitro and in vivo. In in vitro methods, diversification is often created by error-prone replication of DNA. Although used extensively, it needs many rounds of transformation for the selection step that is time-consuming and requires hosts with high transformation efficiencies. With the emergence of novel gene targeting and editing tools, in vivo mutagenesis methods are being used more prominently allowing continuous directed evolution to be performed more conveniently [5]. However, existing tools for the in vivo diversification lack some important features of a perfect mutagenic tool:
- The generated mutations need to span a broad sequence length to produce a great pool of diversified sequences to have high numbers of variant functional proteins. As a result, it is impractical to use targeted mutagenic systems for such diversification. For instance, systems using dCas9 anchored to a specific mutator domain [6] generate mutations at a specific locus in the sequence, and exploiting them for long stretches of the sequence is not convenient as it would require hundreds of guide RNAs. Additionally, a perfect in vivo mutagenic tool should be able to produce every kind of mutation without having a bias towards a specific type of mutation.
- Among the non-targeted diversification systems, the most common issue is that they produce mutations in the host genome and vector backbones in addition to the desired sequence which can reduce the efficiency of the selection process due to additional toxicity and the emergence of “cheaters” (variants that show improved selection due to a compensatory mutation in the genome or vector sequence, rather than the targeted protein). As an example, the mutagenic plasmid used in phage-assisted continuous evolution (PACE) technology [7] encodes for a mutant E. coli DNA Pol III proofreading domain resulting in the mutagenesis to occur in the E. coli genome as well as the vector carrying the gene of interest intended to be evolved. Another elegant tool uses an error-prone DNA polymerase which only recognizes the ColE1 origin of replication [8]. This system exhibits fewer off-target mutations, but off-target mutations are still created in the vector. Another disadvantage of this tool is that it performs only in E. coli, and more mutations are observed in the vicinity of ColE1 Ori [9].
Ideally, the mutagenesis rate should be comparable to the high-level rate of in vitro error-prone PCR [5].
To address the first two issues, tools based on the orthogonal RNA polymerases of phase T7 (T7RNAP) have been developed recently [10–13]. Mutagenesis is performed by a mutator protein e.g. base deaminase (Figure 1) linked to the polymerase and, thanks to T7RNAP’s specificity towards the T7 promoter, it is possible to target the mutagenesis to a sequence flanked by T7 promoter and terminator. The high processivity of the T7RNAP allows for mutagenesis to occur over a long sequence of DNA without showing bias to the distance from the promoter. Four methods based on this concept have been developed to make mutagenesis in E. coli: MutaT7 [10], T7-DIVA [11], eMutaT7 [12] and TRIDENT [13]. In the first reported work, MutaT7, a cytosine deaminase (rApo1) was linked to the T7RNAP and a terminator array was used to define the mutagenesis boundary. Although there was a huge increase in on-target mutagenesis (66%) compared to MP6 mutagenic plasmid (0%), some off-target mutations were also detected. The limited mutation spectrum and low rate of mutagenesis (0.34 mutations day−1 kb−1) were the highlighted disadvantages of this system.
The eMutaT7 tool was developed to improve the low mutation rate of MutaT7 by replacing the rApo1 with a more efficient cytosine deaminase (pmCDA1) and inhibition of uracil–DNA glycosylase (UNG) in the host strain. As a result, eMutaT7 could generate mutations with a rate of ∼3.7 mutations day−1 kb−1.
In the subsequent T7-DIVA tool, the authors tried to improve the method in two aspects. To increase the mutagenesis spectrum, apart from three different cytosine deaminases, an adenosine deaminases TadA* was linked to T7RNAP. In addition, to reduce the off-target mutagenesis of MutaT7, dCas9 was used to make a physical barrier blocking the T7RNAP progress to set the mutation boundary. One problem spotted for MutaT7 and T7-DIVA is that mutations are likely to happen in the non-template strand. This is due to the inaccessibility of the template strand due to interaction with the polymerase.
When our work in the wet lab was already started, the TRIDENT system was published [13] with the aim to improve the tunability and mutational diversity generated by the T7RNAP. In this case, by adding DNA repairing factors to the mutagenesis machinery, TRIDENT was successful in generating transitions and, remarkably, T to G transversions.
 Figure 1. Adenine or cytosine deamination as a source of mutation. Cytosine deaminases produce uridine in DNA (A), that pairs with adenosine (B) and leads to a C -> T transition. Adenine deaminases produce inosine in DNA (C), that pairs with cytidine (D) and leads to an A -> G transition.
Figure 1. Adenine or cytosine deamination as a source of mutation. Cytosine deaminases produce uridine in DNA (A), that pairs with adenosine (B) and leads to a C -> T transition. Adenine deaminases produce inosine in DNA (C), that pairs with cytidine (D) and leads to an A -> G transition.
Our project: Evolution.T7
Here, in our project named Evolution.T7, we tried to improve two of the mentioned shortcomings of the current tools. As pointed out, the mutagenesis in the T7RNAP-based systems is biased mostly to the non-template strand. To address this, we designed the target sequence putting the T7 promoter upstream and downstream of the gene to have polymerases moving in both directions and generating mutations in both strands. This approach has been tried by Moore et al. [10] but they found out that this would lead to the accumulation of A:T pairs along the gene. Also, we assumed that owing to the high processivity of T7RNAP, often there are active transcription machinery moving in both directions and thus, there is a high probability of collision resulting in the reduced processivity of both complexes.
This phenomenon can lead to the accumulation of mutations on one DNA strand in one part of the gene and more mutations on the other strand in the other part. To bypass this, our innovative design takes advantage of a mutant T7RNAP that has higher specificity to an altered T7 promoter sequence [14]. The canonical and mutant T7 promoters are installed upstream and downstream of the gene, respectively (Figure 2). The wild-type T7RNAP expression is induced by tetracycline, while the mutant T7RNAP is induced by arabinose. By adding the inducers sequentially, mutations are generated all over the gene sequence and the collision of mutagenesis complexes is prevented.
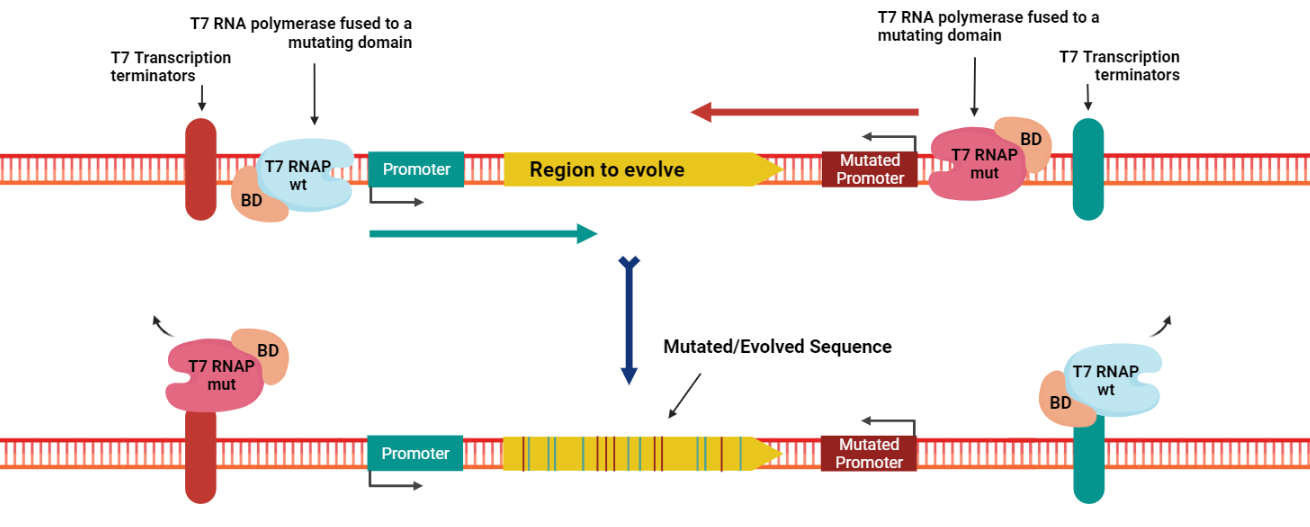
Figure 2. The principle of Evolution.T7 tool.
We also aimed to enhance the mutation rate and diversity. The activity of some base deaminases is affected by the context in which the target base is. For instance, APOBEC1 disfavours the deamination of the cytosine if the neighbouring base is guanine [15]. To overcome these limitations, our tool expands the panoply of base deaminases by including, apart from the ones used in previously described methods (AID, pmCDA1, rAPOBEC1, TadA*), three of their potent variants ABE8.20m [16], evoAPOBEC1-BE4max [17], evoCDA1-BE4max [17]. In addition to higher efficiency, these variants generate mutations without being affected by the base context of the substrate, resulting in higher target compatibility.
The performances of our system were evaluated by various means like the impairment of the fluorescence of fluorescent proteins and the NGS sequencing of the library of mutants produced by each mutator domain (see the "Engineering" page of this wiki). In addition, we constructed a deterministic model of the genetic diversity that our system can produce as a function of time and of the mutagens used (see the "Modeling" page of this wiki). This model allowed us to adjust the experimental setup.
Moreover, as a proof of concept, we applied our Evolution.T7 tool on an antibiotic resistance gene (the AmpR β-lactamase) and transcription factor-based biosensor (LacI) as their alterations can be directly observed and represent a fast and simple screening method (see the "Proof of Concept" and "Improvement" pages of this wiki).
Evolution.T7 is a highly efficient and easy-to-use targeted directed evolution tool, that has the potential to be further developed to increase its modularity and expand the nature of mutator domains beyond the cytosine and adenine deaminase (see the "Implementation" page of this wiki). Moreover, Evolution.T7 is compatible with a continuous operation mode in a bioreactor, thus making the diversification, selection and transmission cycle more productive.
References
[1] Morrison MS, Podracky CJ, Liu DR. The developing toolkit of continuous directed evolution. Nature Chemical Biology (2020) 16: 610–619.
[2] d’Oelsnitz S, Ellington A. Continuous directed evolution for strain and protein engineering. Current Opinion in Biotechnology (2018) 53: 158–163.
[3] Taylor ND, Garruss AS, Moretti R, Chan S, Arbing MA, Cascio D, Rogers JK, Isaacs FJ, Kosuri S, Baker D, Fields S, Church GM, Raman S. Engineering an allosteric transcription factor to respond to new ligands. Nature Methods (2016) 13: 177–183.
[4] Grazon C, Baer RC, Kuzmanović U, Nguyen T, Chen M, Zamani M, Chern M, Aquino P, Zhang X, Lecommandoux S, Fan A, Cabodi M, Klapperich C, Grinstaff MW, Dennis AM, Galagan JE. A progesterone biosensor derived from microbial screening. Nature Communications (2020) 11: 1276.
[5] Wang Y, Xue P, Cao M, Yu T, Lane ST, Zhao H. Directed evolution: methodologies and applications. Chemical Reviews2021; doi 10.1021/acs.chemrev.1c00260.
[6] Halperin SO, Tou CJ, Wong EB, Modavi C, Schaffer DV, Dueber JE. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature (2018) 560: 248–252.
[7] Esvelt KM, Carlson JC, Liu DR. A System for the Continuous Directed Evolution of Biomolecules. Nature (2011) 472: 499–503.
[8] Camps M, Naukkarinen J, Johnson BP, Loeb LA. Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proceedings of the National Academy of Sciences of the United States of America (2003) 100: 9727–9732.
[9] Troll C, Yoder J, Alexander D, Hernández J, Loh Y, Camps M. The mutagenic footprint of low-fidelity Pol I ColE1 plasmid replication in E. coli reveals an extensive interplay between Pol I and Pol III. Current Genetics (2014) 60: 123–134.
[10] Moore CL, Papa LJ, Shoulders MD. A processive protein chimera introduces mutations across defined DNA regions in vivo. Journal of the American Chemical Society (2018) 140: 11560–11564.
[11] Álvarez B, Mencía M, de Lorenzo V, Fernández LÁ. In vivo diversification of target genomic sites using processive base deaminase fusions blocked by dCas9. Nature Communications (2020) 11: 6436.
[12] Park H, Kim S. Gene-specific mutagenesis enables rapid continuous evolution of enzymes in vivo. Nucleic Acids Research (2021) 49: e32.
[13] Cravens A, Jamil OK, Kong D, Sockolosky JT, Smolke CD. Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nature Communications (2021) 12: 1579.
[14] Meyer AJ, Ellefson JW, Ellington AD. Directed evolution of a panel of orthogonal T7 RNA polymerase variants for in vivo or in vitro synthetic circuitry. ACS synthetic biology (2015) 4: 1070–1076.
[15] Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature (2016) 533: 420–424.
[16] Gaudelli NM, Lam DK, Rees HA, Solá-Esteves NM, Barrera LA, Born DA, Edwards A, Gehrke JM, Lee S-J, Liquori AJ, Murray R, Packer MS, Rinaldi C, Slaymaker IM, Yen J, Young LE, Ciaramella G. Directed evolution of adenine base editors with increased activity and therapeutic application. Nature Biotechnology (2020) 38: 892–900.
[17] Thuronyi BW, Koblan LW, Levy JM, Yeh W-H, Zheng C, Newby GA, Wilson C, Bhaumik M, Shubina-Oleinik O, Holt JR, Liu DR. Continuous evolution of base editors with expanded target compatibility and improved activity. Nature Biotechnology (2019) 37: 1070–1079.












© 2021 • iGEM Evry Paris-Saclay