Team:CCA San Diego/Results

Overview
Our results consist of our DNA and protein gel verification of our Gibson assembly and lithium acetate transformation into yeast. We also present the results of extracting animal collagen in order to work on our proposed implementation of collagen hydrogels.
Goals:
- Have colonies grow on a selective ampicillin (+) plate and show that our plasmid is present in a gel next to our linearized backbone and correctly sequenced.
- Grow yeast colonies on a SD dropout URA3(-) plate and show PCR verification of expression via amplifying overhangs and correct protein expression on an SDS-PAGE.
- Obtain triple helical collagen from porcine skin for use in collagen gels.
Before diving in, we want to note that we will be mostly presenting our successful results and more documentation of our trials can be found on our Engineering Success page.
Expression
DNA Preparation
To create a plasmid that could be cloned in yeast cells, several linear fragments had to be prepared, as the desired scl2 construct (BBa_K3956014) by itself was too complicated to be prepared by custom synthesis. Throughout the optimization of these fragment designs with 80-bp overhangs to each other, the linearized plasmid backbone consisting of the pRS4116 plasmid (with the lacZalpha and f1ori regions removed) was kept constant. Figure 1.1 compares the lengths of the amplified, linearized backbone to the full pRS4116 plasmid, with the linearized fragment in lane 1 slightly shorter, implying successful PCR. The presence of this linear backbone fragment was further verified after a DpnI digest was conducted to remove any methylated DNA still part of a circular plasmid backbone (Figure 1.2). As the PCR product only has one visible band around the desired length, 3754 bp, in comparison to the same concentration of plasmid backbone, it can be concluded that the DpnI was successful.


Throughout the experiment three construct designs were tested - two trials broke the construct into three overlapping segments, while one was able to break it into two overlapping segments. The fragments were ordered from IDT, amplified, and purified for optimal Gibson Assembly. The compiled results of the amounts of DNA used in Gibson and Yeast are listed below. All the DNA was used in assembly after using a DNA cleanup kit and measuring the concentration with a NanoDrop spectrophotometer.
Gibson Assembly and Transformation of E. coli
Our fourth Gibson Assembly was successful after assembling a plasmid with three fragments - the truncated pRS4116 backbone, and the scl2 construct broken up into two overlapping segments. Figure 1.3 shows that a miniprep from an efficacious colony is slightly larger than the linearized pRS4116 backbone, implying a construct the correct length of 1390 bp was inserted (band in lane GIB is slightly larger than pRS). In addition, the miniprep was amplified with primers for the inserted scl2 fragment, which showed a successful amplicon around of expected length 1396 bp.

However, upon sequencing, the plasmid was shown to match the same base pairs as our DNA design backbone but not our construct, which prevented us from transforming our assembled 5144 bp pRS4116-His-scl2 fragment into yeast.
In our next iteration, the same DNA fragments were used and colonies grew on the appropriate Amp + plate. New primers were designed in order to amplify overlapping segments where successful ligation should occur in Gibson and yeast assembly. The two primers were designed to amplify a 678 bp fragment where our construct and backbone were ligated, and we were able to verify that in the 2-fragment Gibson Assembly, the primers did amplify the overhangs of the correct length, implying successful ligation (Figure 1.7).


These colonies (shown in the last row of Figure 3.2) were sequenced with primers around the overlapping segments of the construct and backbone. Again, while the backbone was successfully sequenced, the ORF was not successfully sequenced. Although we transformed this miniprep into yeast and saw successful results, the sequencing results were not supportive of a successful assembly. Overall, as the overhangs were successfully amplified and the correct length on the gel, and the transformed bacterial and yeast colonies with the assembled plasmid grew successfully on both Amp+ and Ura- plates, we believe that the assembly was successful.

Throughout all of our trials, we recorded the amount of each fragment used in the Gibson Assembly, aiming to use between .2-1.0 pmol as recommended by NEB. Our most successful trial used the middle range - 0.624 pmol of DNA, with an approximate 2.5:1 ratio of inserts to backbone (Table 1).
Lithium Acetate Gap Repair Cloning
There were a total of eight yeast assemblies with LiOAc transformation trials. For the first seven trials, we utilized a standard LiOAc/PEG/H2O protocol, but for the eighth trial of yeast assembly, we used a new procedure with additional solvents: DMSO and TE buffer.
For our seventh iteration before we modified our protocol, we transformed the successful plasmid from Gibson assembly into yeast as well as directly transforming linear fragments for gap-repair cloning. We noticed that there weren't many colonies that grew in comparison to our pRS plasmid, but more colonies than our no DNA control plate. In order to verify that transformation was successful, we used genomic yeast extraction with ethanol precipitation before running a PCR with the same primers amplifying overhangs as we did in Gibson. However, no bands were visible when run in a gel, which upon reflection may be due an intact cell wall and failure of the genomic extraction rather than the assembly itself, as we used a different protocol for our last trial that showed successful bands.
In our last trial, we showed the most promising results in terms of the number of colonies that grew on selective media. Figure 3.2 shows that the yeast transformation of our plasmid backbone (as a positive control as it contains a URA3 gene) and Gibson Miniprep grew three colonies, while the negative control had one colony and a two-fragment yeast assembly had no large colonies. Because the miniprep plasmid from our last successful Gibson showed large colonies, we again attempted to extract the genomic DNA to run a PCR.

To isolate the DNA, we used a new protocol similar to a bacterial plasmid miniprep, and the PCR (Figure 1.8) showed one successful lane from the 2-fragment Gibson assembly of the ligated region of the plasmid around 678 bp. Interestingly, both the lane from the Gibson and the other controls from the yeast assembly, pRS4116, and no DNA transformation had strong amplicons around 1,500 bp. As the primers designed were unique in our plasmid, it may be possible, but highly unlikely, that there is a very similar genomic region within endogenous yeast chromosomes that match both primers. Alternatively, a larger sequence may have been inserted within the construct through the gap-repair process or the Taq Polymerase in the PCR reaction ran on to amplify an entire section of the plasmid due to statistical mispriming of another primer.
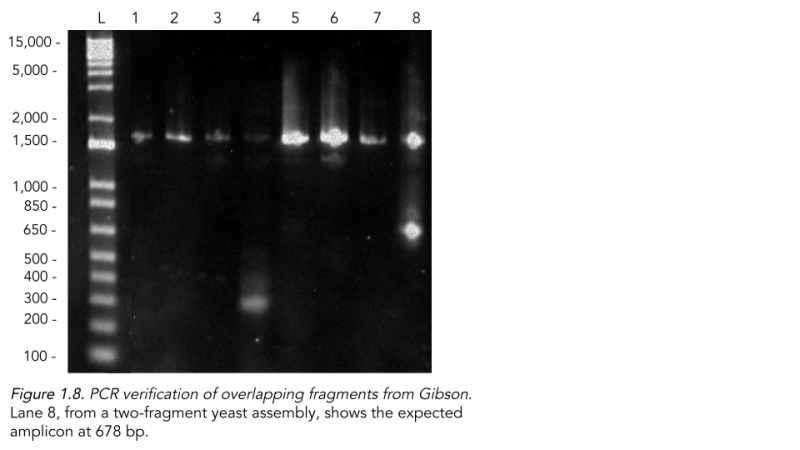
We again recorded the amount of DNA used in our Yeast Assembly (Table 2). As there were no definite guidelines on how much DNA to use, although protocols advise against excessive volume, the total pmol of DNA used varied more widely compared to the Gibson. Our last successful trial used a total of 1.810 pmol of DNA (second trial of the Two-Fragment Construct), which indicates that the amount of DNA does not necessarily lead to a successful assembly. As there was a successful amplicon around 678 bp, we proceeded in our attempts to verify the actual expression of the scl2 collagen protein through an SDS-PAGE.
Verification of scl2 expression
The verification that the DNA had been transformed into the chassis after they grew on selective media was done in several sequential steps. First, for Gibson assembly, the plasmid was extracted via miniprep and run in a gel alongside a linearized pRS4116 fragment to test if the plasmid had increased in size. To verify that the construct was correctly inserted, primers were designed to amplify the overlapping regions on both sides of the construct, with PCR products 678 bp and 713 bp in length, respectively. This was run in a gel to test if the overhangs were assembled correctly.
Finally, to verify the expression of the actual protein, several SDS-PAGE trials were run to varying degrees of success, finally obtaining proper cell wall digestion with Zymolyase to isolate protein lysates, as well as correct staining of the gel with Coomassie. The final protein gel shows many bands as expected, including the Zymolyase enzyme around 52 kDa, but not the expected construct around 42 kDa. As there is not much time left to submit results, we may have to stop here. However, we do have some promising DNA gel evidence that the construct was inserted into the yeast, and next steps with Western blotting for the His-Tag or changing promoters or chassis could verify our construct design.

Discussion
In our lab work to craft CollaGene, we performed several Gibson assembly trials, lithium acetate transformations, and verification with both protein and DNA gels. Although we were unable to show scl2 expression within our chassis through a clear band on an SDS-PAGE, we were able to show that our designed plasmid was successfully inserted through colony PCR overhang length verification.
Prior to beginning our project, we were aware that it would be difficult to express collagen due to the repetitive nature of the collagen-like domain, with 80 Gly-Xaa-Yaa repeats. The countless amount of trials that was required to assemble the plasmid in bacteria and yeast may have been a reflection of this, as working with repetitive sequences is prone to error, especially during PCR which can form hairpin loops and dissociation of the Taq polymerase from the template during amplification, which leads to further “mega primers” that generate unwanted amplicons (Riet et. al, 2017). As our team amplified our gBlocks and genes before Gibson and Yeast assembly, it is possible that the repetitive nature of the scl2 construct prevented clear amplification of our desired sequence. To test this, we ran a gel to verify that our PCR amplicons were present on one of our three-fragment designs (Figure 1.4). The third fragment, which contained the most repetitive sequences, was around our suspected length of 445 bp, but contained other bands within the lane as expected from mispriming and repetitive sequences.

Although PCR was not done for every single trial for all three construct fragmentations, it can be inferred that mispriming and incomplete amplification may have occurred and contributed to our null sequencing and expression results.
Throughout our experimentation, the fact that yeast colonies without any DNA transformed into them still grew on selective media, albeit in lower numbers and smaller sizes, was a source of confusion. We believe that the negative controls may still contain uracil from their time spent in the YPD. However, since the colonies were noticeably smaller and lower in number, we believed that our transformation was still successful, and the positive controls and experimental groups would continue to grow in selective media.
Implementation
Overview
When we were considering how to implement the results of our project in a meaningful way, we aimed to create collagen gel, felt, and threads to be used in materials such as sutures and skin grafts. Our implementation results consist of first extracting animal collagen to be then used in our successful gelification of collagen.
Collagen Sourcing
We were ultimately successful in isolating collagen from porcine skin. After testing two different collagen extraction procedures, we found that sodium citrate buffer was most effective in isolating collagen proteins, and we confirmed the presence of the extracted collagen with an SDS page.
The SDS page shows a faint band around 40 kDa in lane in the telopeptide-poor collagen digested with pepsin, indicating structural similarity with our predicted scl2 collagen after proteolysis (Figure 2.1). Further uses of our successfully extracted collagen can be found on our Proof Of Concept page.
Gelification
The engineering process for implementation was a series of gels testing the effects of different variables and reagents. After 8 iterations testing variables from glycerol to citric acid, we optimized the best gelification formula. Previous iterations had demonstrated instant coagulation when collagen gels were procured in neutral pH rather than acidic pH. Glycerol increased the viscosity of the solution while high collagen concentration predictably bolstered total coagulation, but had an observable drop off after 80% concentration.
Our results observed from citric acid and ethanol concentrations had insignificant differences, but based on their respective acidic and neutral pH, we speculated that excess of either would result in zero or lots of coagulation. Specifically, too basic or acidic of a pH would instantly dissolve all current coagulation as observed in the pH trials.
A trend we had noticed was that coagulated collagen would dissipate after prolonged time. We hypothesized that the disappearance of the coagulation was a result of the supernatant dissolving the binded collagen proteins. Thus, our final iteration added this new consideration on top of the mountains of optimization from previous iterations. We condensed the instantly coagulated collagen and then pipetted the supernatant out, replacing it with glutaraldehyde. The result was a small 1cm wide disk of solid collagen gel that acceptably resisted agitation and compression.
Discussion
The result of our implementation experimentation presents strong success in the physicality of making a gel. However, it is questionable whether or not the gel we synthesized is indeed made of collagen, and whether or not the same process can be recreated with Scl2. Unfortunately, we were unable to test both of these questions due to time constraints. But what we can confirm is that the collagen protein source is undeniably pure as demonstrated through SDS pages of the proteins. Thus, our current rationale is that because the collagen source we used was unequivocally collagen protein, the gel is likely to be collagen as well. This is further supported by our experimentation that showed a proportional relationship between collagen concentration and amount of coagulation. Because Scl2 proteins consist of much the same physical structure, we also believe that Scl2 will form a gel with the same gelification procedure.
Future Steps
Our Collagene project has much to improve upon in the future. From improving efficiency of our expression method to actually implementing relevant medical situations, Collagene has a prospective future.
Inducible Promoters
The next step for Collagene is adding inducible promoters, allowing users to toggle the collagen gene expression using various hormones, antibiotics, or even changes in temperature. Studies like Karsenty et al 91 have experimented with various promoters to alter collagen expression and we plan to do the same.
This is especially relevant since our project used a constitutive promoter, which led to an expression of numerous protein bands in our SDS-PAGE. It was difficult to determine whether the protein was expressed with a constitutive promoter, and an inducible promoter will be much easier to regulate collagen expression.
Epidermolysis bullosa
Types of Collagen
3D printing organs
Epidermolysis bullosa (EB) is a disorder that causes skin to become very fragile due to mutations in the production of collagen type VII in humans. The collagen VII is essential in anchoring fibrils in the dermal, which creates epidermal junction that makes our skin the malleable texture. As a possible solution, yeast or bacteria implants or other concoction can be injected into EB patients to enhance collagen production rate.
There are numerous types of collagen that are found for many different organisms, whether that be categories such as mammalian, plant, bacterial, or yeast collagen, or whether that be type I, type II, or type III collagen. Since Collagene is restricted to producing bacterial scl2 collagen in yeast, our next steps are to alter our plasmids to be able to effectively express other types of collagen. Our choice of scl2 was only for its similarity to mammalian collagen, but tackling other types of collagen is our next primary goal.
Printing organs is another possibility with collagen; since 3D printing organs have already been pondered by researchers, implementing collagen will help create a more sturdy, realistic organ because it helps create tendons and ligaments. Studies like Lee et. al 19 has already made efforts to bioprint the human heart and numerous other studies show that collage would be an effective biofuel.
References
Riet J, Ramos LRV, Lewis RV, Marins LF. Improving the PCR protocol to amplify a repetitive DNA sequence. Genet Mol Res. 2017 Sep 21;16(3). doi: 10.4238/gmr16039796. PMID: 28973773.
Karsenty, G., & de Crombrugghe, B. (1991). Conservation of binding sites for regulatory factors in the coordinately expressed α1 (I) and α2 (I) collagen promoters. Biochemical and biophysical research communications, 177(1), 538-544.
Lee, A. R. H. A., Hudson, A. R., Shiwarski, D. J., Tashman, J. W., Hinton, T. J., Yerneni, S., ... & Feinberg, A. W. (2019). 3D bioprinting of collagen to rebuild components of the human heart. Science, 365(6452), 482-487.








