Team:CCA San Diego/Design

Collagen is the most abundant protein in many mammalian organisms and it is a proven biomaterial. However, the current source of collagen is from animal skin, bones, tendons, and cartilage, meaning mass producing collagen would require mass slaughter of animals. It is an unethical, unsustainable, and un-environmental method of extraction. Our goal is to create an alternative method for mass producing collagen to be used in various biomedical applications. We chose to express a bacterial collagen-like protein, Scl, in yeast. Our process can be broken into three parts: expression, purification, and implementation, with each part playing a crucial role in our project outline. The primary component of our expression part is creating our plasmid; we put a lot of time and consideration into the various aspects to replicate human collagen. Our main concern with the purification part of our project is purifying the bacterial collagen from host proteins with an environmentally friendly method, which led us to choose pepsin for the proteolysis. Finally, the most important aspect of our project is the implementation. Our collagen is versatile—collagen thread, gel and felt—and we have proposed to use it in various applications.
Expression
Designing the Constructs
Scl, streptococcal collagen-like protein, is a bacterial collagen that has been well-documented in recent years. The key distinction between bacterial and mammalian collagen is the hydroxyproline post-translational mechanisms: mammalian collagen requires this complex process whereas bacterial collagen does not. Because of these hydroxyproline specifications, dealing with mammalian collagen is extremely difficult to work with and mass produce. Thus, Scl2 was the bacterial collagen source of our choice. Past studies show that Scl2 has similar enough properties to human collagen to be used as biomaterials while also avoiding the lengthy processes of hydroxylation.
Digging deeper into Scl2, most types of scl proteins, including Scl2, are naturally expressed in the human pathogen, Streptococcus pyogenes, also known as Group A streptococcal (GAS). The Scl proteins are virulence factors of S. pyogenes and contribute to the formation of biofilm adhesion and infection of different species (Squeglia et al., 14). The Scl2 gene itself is made of several parts: the V domain, the triple helical CL domain, and the tail. When building the plasmid to express Scl2, we had to add a 5’UTR in front of the codon to make sure the triple helical structure was produced.

The plasmid host of our choice was Saccharomyces cerevisiae, a type of yeast. Though the idea of expressing bacterial collagen in yeast may seem strange, past studies have shown that expressing bacterial collagen in E.coli is problematic and the yield is often too low. Our team decided that expressing Scl2 through yeast will be a novel experiment that also gets higher yields than bacterial expression. Though there has been significant effort in efficiently producing human and animal collagen through yeast, there have been no studies using yeast for bacterial collagen. Previous literature also suggests using Scl2 in yeast for increased chain length (An et al., 14).
Choosing the Right Promoters and Terminators
Choosing the optimal promoters and terminators are crucial in initiating expression of our Scl2 construct, especially since we chose to express a bacterial protein in a eukaryotic organism. Although promoters are well-known to initiate enzyme docking and expression of a recombinant gene, the careful consideration of terminators is often neglected due to a common assumption that the sequence after the gene and the poly-A-tail folding is unimportant. However, a study by Curran et. al, 2013, challenged this assumption by demonstrating a significantly (3.5x) higher expression of fluorescent proteins when high-capacity yeast terminators were used. Because of this, we decided to choose a high-capacity natural yeast terminator correlated with the strongest expression from their paper, PRM9, which was first documented by Miller et. al, 2011.
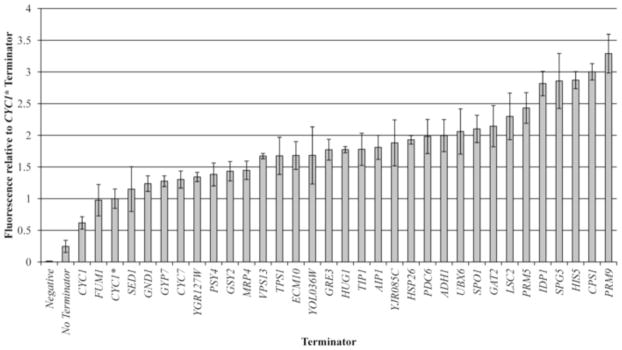
To choose our promoter, we had several criteria we wanted to fulfill: the promoter should exhibit higher protein expression compared to commonly used promoters, and it should be constitutive and synthetic. Natural promoters with a high variance across different experiments have a lower predictability and may make their heterologous pathways unfavorable (Decoene et. al, 2019). In other words, synthetic promoters may be optimal for genetic engineering purposes compared to natural promoters, as it reduces the risk of off-target binding when targeting the expression of recombinant constructs. After searching the literature on the topic, we found that the synthetic TEF1-derived promoters documented by Decoene et. al, 2019, fulfilled all of our criteria. As TEF1 is a well-characterized strong promoter in S. cerevisiae, the study authors randomized their elucidated sequence of the TEF1 minimal core promoter. They also discovered that an upstream activating sequence (UAS) around 100 bp in length dramatically improved fluorescence. The UAS is thought to be where transcription factors may bind to increase docking to the core promoter, where RNA polymerase II initiates transcription. In addition, a 5’UTR sequence usually located after the core promoter was included, as it is known to play a major role in the translation initiation process.
Thus, we chose the most optimal combination of the upstream activating sequence and core promoter developed in their study for use in Collagene. The optimal UAS-core promoter pair was named UASFEC cpTEF-6I, which we kept under the same name when documenting these components as new parts.

Shortening the Plasmid
An important modification in our plasmid design was cutting the LacZ gene and f1 ori. To do this, we conducted a PCR with primers designed to amplify only a fraction of plasmid DNA—excluding the LacZ and f1 ori. Effectively, the final plasmid was shorter than before. This is important because it increases the productivity of our protein expression, a key point in our goal of mass production. The smaller size also gives the benefit of easier transformation into a host. Larger molecules typically struggle more than smaller ones when entering the membrane of a host cell. Prior to using our shortened linearized backbone, we made sure to remove any template, full-length backbones with a DpnI digest. If the plasmid template is not cleaved before assembly, the transformed microorganisms might still contain all the selective auxotrophic or antibiotic resistance genes without our gene of interest.
Dpn1 is an enzyme that only cleaves methylated plasmid DNA. Template plasmids are derived from a dam+ E. coli strain and will have methylated adenines in any GATC sequence on the plasmid. Thus, the template plasmid will be cleaved by DpnI leaving behind the amplified PCR fragment which can be verified by sequencing.
Putting It All Together
We struggled with ordering our plasmid as IDT and Twist were unable to synthesize the full sequence. The problem stemmed from repeating triplets of nucleotides in the CL-domain of the Scl2 gene. Unfortunately, these repeating sequences are necessary for the triple helical structure of Scl2. Our team adapted to the problem and ordered the full sequence in multiple pieces.
Our first design was buying the sequence in over 10 segments: promoter, His-tags and globular domain, enzyme cut site, 7 Scl2 segments, and the terminator. Assembling this would have been extremely difficult and highly susceptible to error. Instead, we ordered it in three sequences and amplified them with 6 interweaving primers. Each segment was amplified with only its accompanying forward and reverse primer. Fragment 1 contained gblock, cpTEF promoter, and 5’UTR. Fragment 2 contained Scl2 encoding DNA. Fragment 3 finished with the PRM9 terminator. Notably, we did not need to delete the secretory signal which had the result of maintaining the integrity of our DNA sequence. With the primers and fragments prepared, DNA assembly was the next step.

Assemblies in Parallel
Since our project revolves around expressing the Scl2 gene through yeast, one of our methods for transforming the S. Cerevisiae is yeast assembly, also known as LiOAc (lithium acetate) transformation. Yeast assembly allows users to order recombinant plasmids inside the yeast in a single step, as opposed to other methods which often require the plasmid to be fully ordered before being inserted and expressed by yeast. By doing the entire process in the yeast, the process takes much less steps and therefore is more error-proof.
The other method we chose was Gibson assembly, a procedure that allows for the binding of multiple linear DNA fragments and avoids common errors associated with assembling plasmids. The Gibson assembly is standard for assembling plasmids for E. coli, but can be beneficial for yeast plasmid assembly by successfully conjoining plasmid parts. Gibson will be used to assemble the backbone and the Scl2 plasmid part. Thus, the Gibson assembly essentially serves as our control assembly method. Due to the codon optimization of the yeast, the transformation may be less efficient because the E. coli is not trained to use the proteins.
As mentioned, the yeast assembly and Gibson assembly were done in parallel to see which process was more efficient. Both methods are known to be more effective than the standard restriction enzyme-based cloning method.
Purification
His-Tag Purification and Modified TCA Extraction
The His-tag and modified TCA extraction are useful tools for protein purification. Since the His-tag and Modified TCA extraction will achieve the same purpose, we will do them in parallel to see which method produces better results. Before deciding on modified TCA extraction, we initially planned to use glass bead protein extraction, then considered traditional TCA extraction, and then considered the extraction method in the Peng et al. 2014 study. A problem with collagen lies in its denaturation slightly above body temperature. Xu et al. 2010 showed that the stability at pH 2.2 of S. pyogenes collagen was 25.7 C. Therefore, the TCA extraction has to be below 28 degrees, which is why we are using a modified TCA extraction which requires less time, so excessive protein degradation is avoided.
Protelysis
We used the Peng et al. 2014 study to compare and contrast the different enzymes that could be used for proteolysis. Proteolysis is used to remove contaminating host proteins to produce pure bacterial collagen proteins. In the study and figure, trypsin and chymotrypsin were shown to be ineffective while pepsin was shown to be the best enzyme for removing remaining host proteins.
Papain was found to be effective as well. Papain is a natural enzyme derived from papayas that is used in the leather industry to hydrolyze collagen peptide bonds and has a similar function as pepsin. Papain has the added benefit of being a non-animal protease, allowing for a completely non-animal purification system. However, pepsin also hydrolyzes collagen peptide bonds, is further studied, is used more often to make collagen hydrogels, and is more economical for our purposes than papain, sealing our decision to use pepsin for proteolysis.
Implementation
Choosing our Implementation
When considering how to use the collagen we had created in an effective and applicable way, there were a myriad of concerns to address: could we be sure that our collagen products would be an improvement from the status quo? If not, how would we make it that way? How difficult would it be to guarantee structural integrity of the products—and if we could do so, what would the tradeoff be?
To answer these questions, we consulted numerous professors (INSERT HYPERLINK), who—using their experience with collagen production and utilisation—helped us determine what would be feasible, given our resources and materials.
Overall, the three implementations we decided on—thread, gel, and felt—displayed the potential of our yeast-expressed collagen the best. While all extremely different in their manufacturing processes and applications, the collagen substances were created from the same material—demonstrating collagen’s versatility and possibilities.

Coagulation
To make our collagen usable in practical fields, it needed to contain a functional triple helical structure—hence the need for coagulation. Collagen molecules are in their most stable, precipitated state at a neutral pH rather than an acidic or basic pH (Hayashi et al 73). We used the solubility of collagen in an acidic solution to form our collagen stock solution and neutralized the solution when coagulation was necessary. We did so by extruding the collagen into a buffered solution of polyethylene glycol with a pH of 7.55, allowing the collagen to stabilise (Cavallaro et al 94).
Thread
When designing our thread, our primary reference was “Collagen Fabrics as Biomaterials,” written in 1994 regarding the derivation of collagen, manufacture of thread, and production of fabrics using unique weaving patterns for the purpose of wound treatment (Cavallaro et al 94). To modify the procedures provided according to the properties and structure of our yeast-expressed collagen—and materials available to us—we contacted many experts to give us feedback on our ideas and potentially offer new ones.
One of the professors we spoke with was Dr. Samuel Hudson of Wilson College, an expert in textile engineering. He helped us develop effective fiber testing procedures in addition to manufacturing methods that would ensure the structural stability of our thread. He also provided us with numerous options for crosslinking reagents in addition to their properties and hazards.
We also consulted Dr. John F. Cavallaro, an author of the paper we had been using as reference. He helped us solidify our procedures, determine the final implementations of our project, and gave us ideas regarding how we might source the collagen we needed. At first, we planned on just creating sutures from our collagen, but Dr. Cavallaro gave us ideas for numerous novel applications that would properly demonstrate the versatility and practicality of our product—rather than simply something that had previously been researched or discovered.
Afterward, we contacted Dr. Oded Shoseyov of the Hebrew University of Jerusalem who gave us remarkable insight on crosslinking reagents, our wet-spinning apparatus, and collagen sourcing and properties. Initially, we had planned on using UV crosslinking to strengthen the collagen, but after learning from Dr.Shoseyov that it might be too weak, we came to the conclusion that glutaraldehyde would be a better alternative.
When it came to the issue of finding a wet-spinning machine for manufacturing thread, we were inspired by a previous iGEM project by GreatBay_SZ in 2019, that successfully created a synthetic spider silk with a 3D-printed spinning machine. We decided to use their design because of their success with similarly synthetically produced biomaterials. We were able to print the GreatBay machine and made specific modifications for use with collagen.

In essence, the final wet-spinning process involves the following steps:
- Collagen is extruded into a coagulation bath, allowing it to stabilise - consequently forming a continuous thread.
- The thread is then soaked in a rinse, bath, which helps it to maintain its constant pH.
- It is passed through an isopropanol bath, which partially dehydrates it.
- It is drawn over a series of pulleys and dried under tension.
- It is then cross-linked with glutaraldehyde.
Gel and Felt
Similar to the manufacturing process of the thread, the primary issue with both the gel and the felt was that of structural integrity and coagulation. For the process of making gels, we looked to Ho et. al ,1996, and their method of collagen gelification using citric buffers. After trials and trials of iteration after iteration, testing glycerol concentration, collagen concentration, glutaraldehyde presence, pH, and more, we learned new clues each step and drew closer to synthesizing a collagen gel. The gel we successfully produced was made by condensing the coagulated particles in neutral pH and crosslinking agent. Our gel is structurally sound, resisting agitation and reforming after compression.
When designing the felt, we studied the processes of industry felt-making in efforts to better understand the science behind it, thus allowing us to replicate desired aspects to the best of our ability with the materials we had. Generally, felt is made via heat, water, and pressure, with the fibres condensed and pressed together, which permanently interlocks them, creating the matted fabric (Sewport Support Team). Depending on the materials used, the fabric can have different properties, but overall, the most desirable quality of felt is that it involves no knitting or weaving. It serves as one large, flattened piece, which both ensures its structural integrity and greatly minimizes the amount of extra work that goes into production. With our felt, given the restriction of how collagen might be difficult to work with, we decided to follow procedures similar to that of our gel; the differences, however, taking into account the criteria the felt fabric needed to fulfil (retained solidity, thickness, etc.).
References
Hayashi, T., & Nagai, Y. (1973). Effect of pH on the stability of Collagen molecules in solution. The Journal of Biochemistry, 73(5), 999–1006. https://doi.org/10.1093/oxfordjournals.jbchem.a130184.
Ho, H. O., Lin, L. H., & Sheu, M. T. (1997). Characterization of collagen isolation and application of collagen gel as a drug carrier. Journal of Controlled Release, 44(2-3), 103-112. https://doi.org/10.1016/S0168-3659(96)01513-1
Cavallaro, J. F., Kemp, P. D., & Kraus, K. H. (1994). Collagen fabrics as biomaterials. Biotechnology and Bioengineering, 43(8), 781–791. https://doi.org/10.1002/bit.260430813.
https://www.google.com/url?q=https://2019.igem.org/Team:GreatBay_SZ/Hardware&sa=D&source=editors&ust=1629684548117000&usg=AOvVaw0YAos3eazF2gBn-UPFJUK3
Sewport Support Team. (2020, September 10). What is Felt Fabric: Properties, How its Made and Where. Sewport. https://sewport.com/fabrics-directory/felt-fabric.
Miller, C., Schwalb, B., Maier, K., Schulz, D., Dümcke, S., Zacher, B., Mayer, A., Sydow, J., Marcinowski, L., Dölken, L., Martin, D. E., Tresch, A., & Cramer, P. (2011). Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Molecular systems biology, 7, 458. https://doi.org/10.1038/msb.2010.112
Curran, K. A., Karim, A. S., Gupta, A., & Alper, H. S. (2013). Use of expression-enhancing terminators in Saccharomyces cerevisiae to increase mRNA half-life and improve gene expression control for metabolic engineering applications. Metabolic engineering, 19, 88–97. https://doi.org/10.1016/j.ymben.2013.07.001
Peng, Yong Y, et al. “A Simple Cost-Effective Methodology for Large-Scale Purification of Recombinant Non-Animal Collagens.” Applied Microbiology and Biotechnology, U.S. National Library of Medicine, Feb. 2014, www.ncbi.nlm.nih.gov/pmc/articles/PMC3968793/.








