Team:WHU-China/Results

Inhibition Module
GOAL
1. To express and purify PctA, then test its ability to inhibit the growth of P. acnes.
2. To prove that engineered bacteria can secret PctA into the environment, and inhibit the growth of P. acnes.
ACHIEVEMENT
1. The expression vector of PctA was successfully constructed and transfromed into BL21(DE3) strain.
2. Expressed and purified the precursor PctA in BL21(DE3) strain.
3. Tested the antibiotic activity of precursor PctA against P. acnes.
Expression of PctA
In order to get enough protein products for functional test, we chose pET26b(+) as vector and E. coli BL21(DE3) as host cell. We got E. coli DH5ɑ with pET26b(+) from Dr. Chen’s lab and synthesized PctA gene (with restriction site for NdeI at 5’ and XhoI at 3’) from Tsingke Biotechnology company. Plasmid was extracted, and digested with NdeI and XhoI. Correct bands of pET26b(+) vector (little more than 5000 bp) and PctA gene sequence (250-500bp, 289 bp in theory) were observed.

The pET26b(+) vector and PctA gene fragment were mixed and incubated with T4 DNA ligase to form our target plasmid, and then transform DH5α competent cell. After colony PCR and plasmid sequencing verification, the expression vectors were transformed into BL21(DE3) and sequenced again to make sure PctA gene sequence was correct.

The expression of PctA was induced with IPTG in a set of different conditions and checked with SDS-PAGE. Results showed that 11kDa precursor PctA is successfully induced in the bacteria, but no band of 7kDa mature PctA is observed in the culture media, which meant that PctA was not successfully secreted. As shown in Fig.3 (A) and (B), after induction, the pellet of bacteria lysate contained most of the precursor PctA, while the lysate supernatant had a much smaller portion. This indicates that a portion of the expressed precursor PctA was localized at the inclusion body. The molecular weight of PctA seemed to be greater than we expected, this may result from the fact that SDS-PAGE is not suitable for isolating protein under 15kDa, and the relationship between molecular weight and small protein’s mobility can be unreliable.
| lane | volume/ml | IPTG/µl | c(IPTG)/mM | temperature/˚c | Time/h |
|---|---|---|---|---|---|
| 0 | 5 | 0 | 0 | 37.0 | 0 |
| 1 | 5 | 1.5 | 0.3 | 16.0 | 1.0 |
| 2 | 5 | 1.5 | 0.3 | 30.0 | 2.0 |
| 3 | 5 | 1.5 | 0.3 | 37.0 | 3.0 |
| 4 | 5 | 2.5 | 0.5 | 16.0 | 2.0 |
| 5 | 5 | 2.5 | 0.5 | 30.0 | 3.0 |
| 6 | 5 | 2.5 | 0.5 | 37.0 | 1.0 |
| 7 | 5 | 5.0 | 1.0 | 16.0 | 3.0 |
| 8 | 5 | 5.0 | 1.0 | 30.0 | 1.0 |
| 9 | 5 | 5.0 | 1.0 | 37.0 | 2.0 |

Secretion of PctA
We tested the secretion of PctA by Western Blot for this method is highly sensitive and thus fit for identifying protein from culture supernatant even at a low concentration. The result is as follows.

As the result shows, no band of 7kDa mature PctA was found, indicating that the precursor PctA failed to cut off the Sec signal peptide to form the mature PctA, and consequently was unable to get into the culture medium. This may contribute to the formation of inclusive body in lysate pellet. We discussed about the problem of secretion at Engineering.
The function of PctA
After a few attempts of optimizing conditions for PctA induction, we still couldn’t observe the band of 7kDa mature PctA, but we did find that at 30℃, 0.5mM IPTG concentration, 4h we could get most precursor PctA in the supernatant of cell lysate. So we decided to first purify the soluble precursor PctA and test its function.
We used Ni-resin to purify the precursor PctA with His tag from lysate supernatant. In order to get a better resolution at low molecular weight range, the product was analyzed by Tris-Tricine SDS PAGE instead of Tris-glycine SDS PAGE. We tried protocols both according to the instruction of the resin and a method of imidazole gradient purification, but neither method had a satisfying result: the former one left the product impure and the latter one had too little protein of interest in the product.

Then we used the resin-purified product to test the activity of precursor PctA (the later product is too thin to carry out experiment). We mixed the product with the culture of P. acnes and measured the antibiotic activity by the change of OD 600. We also conducted the inhibition zone experiment. The result was as follows.


As the results showed, the OD 600 of P. acnes culture didn’t have a distinct decline after the adding precursor PctA, and in another experiment no inhibition zone is observed around the filter with precursor PctA. So we can conclude that precursor PctA doesn’t have antibiotic activity against P. acnes.
In future experiments, we will try to express and purify mature PctA and test its activity.
Fatty Acids Sensing Module
Goal
1. Design a system that enables the engineered bacteria to sense fatty acids, and measure this ability.
2. Improve the sensitivity towards fatty acids and reduce gene expression leakage of our engineered bacteria, mainly through the following two ways:
A) Retrofit ribosomal binding site.
B) Introduce FadR, a repressor, and carry out directed evolution on it.
Achievements
1. Escherichia coli containing a fatty acid-sensitive promoter was successfully constructed (the strain needs to be further identified) and its ability to sense fatty acids was measured.
2. The RBS was successfully modified to improve the sensitivity to fatty acids of our engineered bacteria.
3. FadR was successfully cloned in E. coli MG1655 and a mutation library was constructed by error-prone PCR.
We synthesized BBa_K3763002(pFadD_Lac-RBS BBa_B0030-GFP-rrnB T1 and T2 terminator) and BBa_K3763003(pFadD_Lac-RBS BBa_K3763000-GFP-rrnB T1 and T2 terminator) from Tsingke Biotechnology company. After the successful transformation of the plasmid into Top10 E. coli cells, we used different concentrations of fatty acids for eGFP induction for different times. The expression level of eGFP was determined by fluorescence detection with a microplate reader, reflecting the performance of the promoter and the RBS.


According to the results, we can find that the performance of our fatty acids-sensitive promoter is improved to a certain extent. Taking 0.125× fatty acid concentration as an example, the fluorescence intensity of BBa_K3763003 is over 11000 after 6 hours, while the promoter of BBa_K3763002 is less than 10000 at three fatty acid concentrations. However, we can see that the optimized promoter still has a high expression leakage and low expression in a short time. However, the experimental results have been able to prove the function of the fatty acids-sensitive promoter, and partially prove the effectiveness and rationality of our design.
We cloned genomic DNA FadR from E. coli MG1655 and digested with HindIII and XbaI. Plasmid (pBAD33) was extracted and digested with HindIII and XbaI as well. Correct bands of pBAD33 vector (around 5000 bp) and FadR gene sequence (1038bp) were observed.
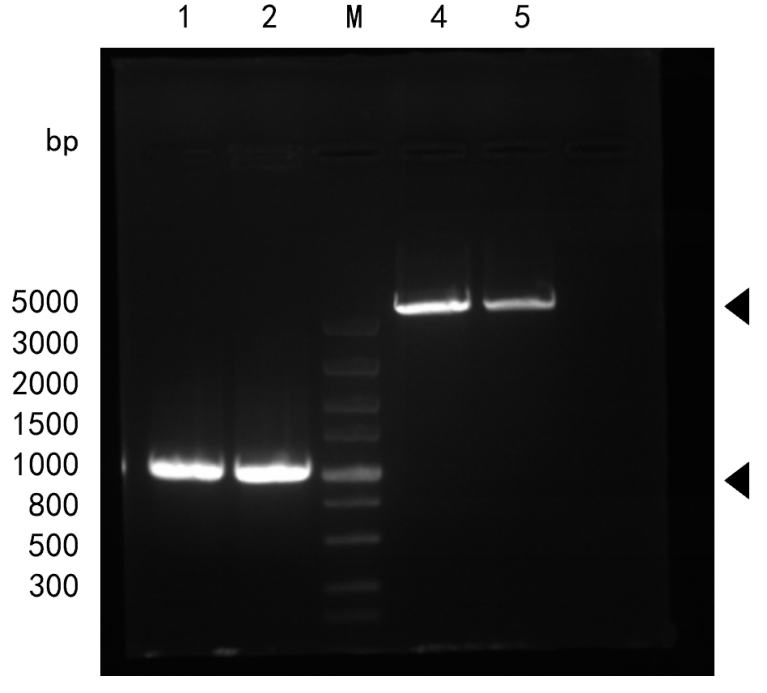
At the same time, we obtained a mutation library of FadR (around 750bp) by error-prone PCR.

The gene sequencing results indicated that we successfully get FadR gene and introduce random nucleotide mutations into the parent sequence qualitatively because there are overlapping peaks.

Our future work will include overexpressing FadR before and after directed evolution in our chassis to reduce the leakage expression of our promotor. Furthermore, we will use the specially designed hardware to perform directed evolution. This will include using the microfluidic hardware to carry out epPCR, setting up cell free expression system to measure the characters of different promotors, and select them using flow cytometry.
Fatty Acid Consumption Module
Goal:
1. To overexpress 2 enzymes in the β-oxidation pathway—fatty acyl-CoA synthetase (FadD) and fatty acyl-CoA dehydrogenase (FadE) —in our engineered bacteria.
2. To equip our engineered bacteria with higher fatty acid decomposition ability by overexpress FadD and FadE.
Achievement:
1. Ni2+ affinity chromatography column and SDS-PAGE proved that we successfully express the target protein with the right molecular weight.
2. The β-oxidation ability of our engineered bacteria was improved.
Experiment:
In order to test whether the overexpression of fatty acyl-CoA synthetase (FadD) and fatty acyl-CoA dehydrogenase (FadE) has a significant effect on improving the fatty acid decomposition ability of our engineered bacteria, we designed the expression vectors of FadD and FadE (Fig1 A,B). FadD is driven by trac promoter, whereas FadE is driven by araBAD promoter. Then we constructed the expression vectors by means of enzyme digestion and ligation and transformed them into E. coli DH5α strains (Figure 1).

After verifying our vector construction by colony PCR and sequencing, we carried out protein expression test. Firstly, we explored the optimal culture temperature, culture time and inducer concentration in the process of FadD and FadE protein expression by orthogonal test. After confirming the optimal expression conditions, we conducted the experiment following the relevant experimental methods of 2019 BNU China—the control group and the experimental group were cultured overnight in LB medium at 37 ℃, and then mixed with the same volume of LB medium containing 400mM fatty acid sodium to make the final concentration of fatty acid sodium 200mM, and then induced them by 1 mM IPTG (FadD) and 0.2% ara respectively (FadE). Samples were taken at 0 h, 2 h and 4 h. The free fatty acid (FFA) content detection kit was used to measure the fatty acid consumption (Fig. 2). The experimental results are shown in the figure below. T-test analysis showed that there was no significant difference in the β-oxidation ability between our engineered bacteria and the original bacteria.

In order to explore the reasons for the failure of the experiment, we verified he protein expression more carefully. After inducing the expression of FadD and FadE proteins, they were purified by Ni2+ affinity chromatography column. After purification, SDS-PAGE results showed that the molecular weight of our target band was about 20 kDa lower than our predicted value. It is speculated that in the engineered strain we used is E. coli DH5α, the endogenous protease system of this strain has not been artificially knocked out, so the overexpressed protein is easy to be degraded. Considering this possibility, we replaced our engineered strain with E. coli BL21 strain. Then we purified the protein by Ni2+ affinity chromatography column and detected it by SDS-PAGE. The results showed that the bands of FadD and FadE protein were in line with our prediction.

While conducting our own experiments to explore the optimal protein expression condition, we sought the help of Ailurus biotechnology and asked them to help us design the best expression vector of FadD protein. Their scalable pipeline enables an ultra-high throughput assay of soluble protein yields under different vectors, with much lower costs than typical lab works.
The construction results of the best FadD protein expression vector are as follows:

Safety module
Goal
1. To obtain a glycine auxotroph.
2. To test the growth rate of auxotroph on additional nutrients and basic media.
Experiment:
We conducted two rounds of gene knockout experiments on hisBCD and four rounds of gene knockout experiments on glyA. Unfortunately, until the end of the experiment process, we have not been able to obtain a successful knockout of nutritionally deficient strain by λ -Red homologous recombination method.
There was no colony growth on the resistance screening plate in the two rounds of hisBCD knockout, indicating that no homologous recombination reaction occurred in the bacteria. So we decided to conduct a single gene knockout. In addition, considering that DH5α has undergone large genetic modifications, MG1655 strain was used as the chassis for gene knockout at the same time in order to avoid the impact of this characteristic on gene knockout.
We repeated four rounds of gene knockout experiments targeting at glyA. Colony growth was found on kanamycin resistance plate in the first and fourth round of experiment with DH5α as the chassis. Primers were designed for PCR amplification of the base sequence near the pre-knockout glyA gene. PCR products were detected by agarose gel electrophoresis to estimate the band size and sequenced to determine whether they contained glyA gene.
The results of first round experiment are as below:

The results of fourth round experiment are as below:

Unfortunately, gene sequencing results indicated that we failed to knock glyA out. Considering that the colony is kanamycin-resistant, it is likely to have off-target effect in the knockout process.
Due to the lack of time, we did not carry out the next round of gene knockout attempt. However, we still believed that off-target effects can be avoided by a suitable primer design. In addition, further research would manage to construct different genetic defects supposing that glyA is important for bacterial growth.













