
We conceptualized and started creating a multi-functional Bacillus subtilis biofilm which detects the pathogen Pseudomonas aeruginosa and in turn activates the expression of a bacteriophage which specifically kills the respective pathogen. Additionally, we developed a kill-switch to prevent the escape of viable, engineered cells from the biofilm.
We here illustrate the process of engineering and highlight the challenges we faced. We implemented the Design-Build-Test-Learn (DBTL) cycle to overcome these challenges and develop new solutions. Practically, we were not able to go past the first iteration of the DBTL cycle, due to the lack of time in the laboratory because of COVID-19 measures. Theoretically, however, we elaborated multiple iterations. In this way, we gradually came closer to our final goal of a safer, self-defending biofilm.
Pathogen sensing
Pathogen Sensing via Transcriptional Regulation
We use two allosteric transcription factors (aTF), which originate from the quorum sensing system of Pseudomonas aeruginosa to create a sensing circuit in Bacillus subtilis sleeper cell (“sleeper state” until being activated). This circuit responds to the presence of P. aeruginosa, a common pathogen, with the production of a bacteriophage which specifically kills P. aeruginosa.
It has been shown that P. aeruginosa cells can be sensed using either the aTFs QscR or LasR.1,2 LasR is a transcription factor, which regulates the expression of genes involved in quorum sensing by binding the promoter PLux.3 QscR is a quorum sensing repressor, which can inhibit the function of LasR. Besides this, QscR is also able to control gene expression directly by binding the promoter PPA1897 thus acting as a transcription factor.4 Both aTFs bind the acyl homoserine lactone (AHL) N-(3-oxododecanoyl)-homoserine lactone (3OC12-HSL) which is a quorum sensing autoinducer native to P. aeruginosa.3,4 The respective aTF and 3OC12-HSL form a complex that binds to its respective inducible promoter. In our circuit, the aTFs are expressed constitutively to control the expression of recA730 via the respective inducible promoter (Figure 1). The protein RecA730 is responsible for the switch from the lysogenic to the lytic cycle of the phage and thus for controlled phage production.5,6
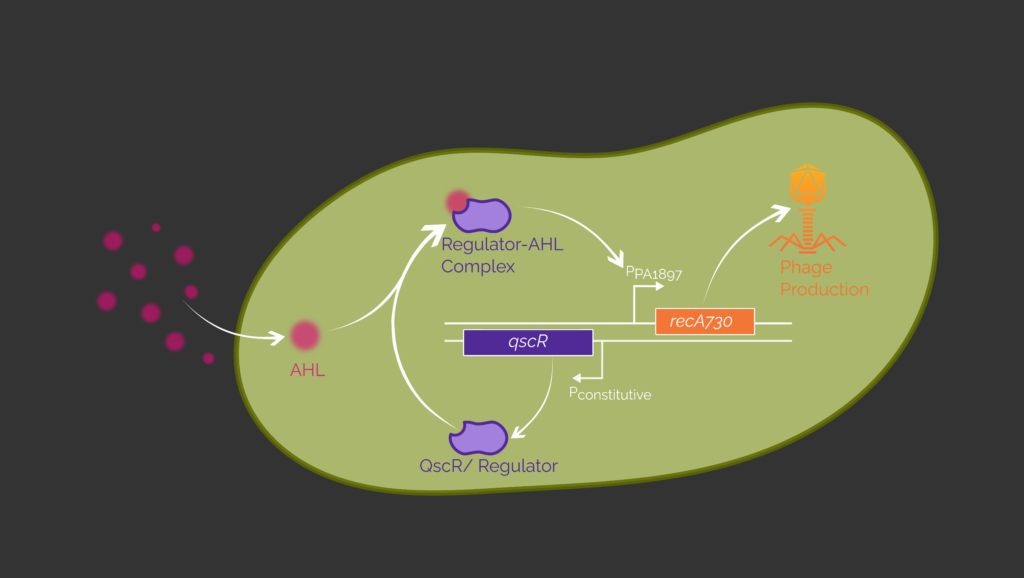
To prove the inducibility of our designed circuit, we exchange recA730 with egfp (Figure 2). If the circuit works properly, we will obtain a green fluorescence signal as an output. Wu et al. (2021) and Saide et al. (2011) have already successfully detected P. aeruginosa using E. coli by expressing egfp in the presence of 3OC12-HSL.1,2
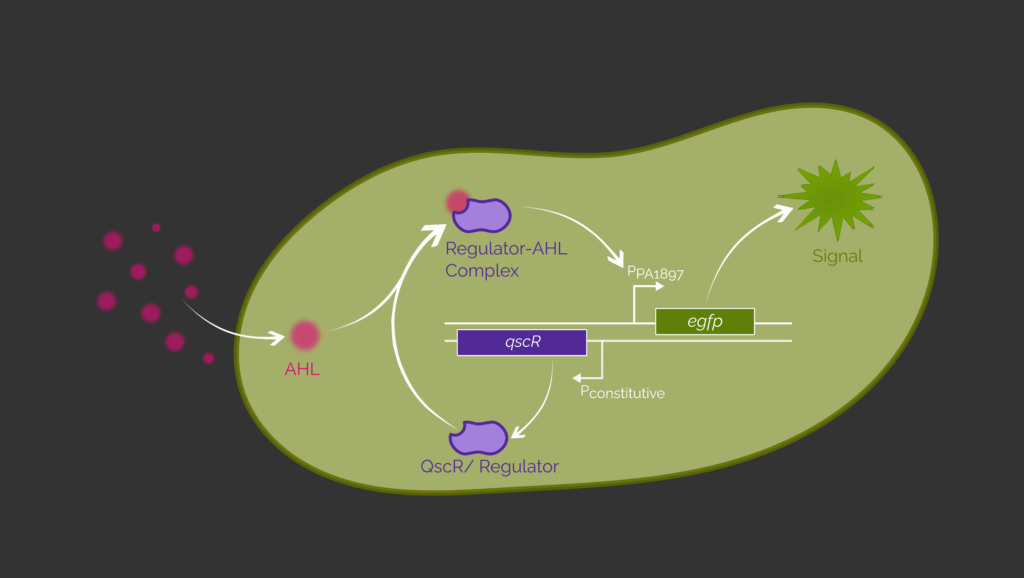
Since this circuit has not yet been evaluated in B. subtilis, we planned to use a DBTL cycle to generate a functional sensing circuit. Although we have been able to assemble the genetic circuit we can not present experimental data of this project part since our time in the lab was limited. Therefore, we performed a theoretical DBTL cycle.
First of all, we want to characterize the constitutive promoter Pveg of B. subtilis. Therefore, we express the red fluorescent protein mKATE2 under its control and measure the fluorescence intensity over time. This way we characterize the used promoter within a fluorescence assay. The possible results are either a measurable fluorescence or none.
- If we measure a fluorescence signal, it means the constitutive promoter Pveg is active and mKATE2 is functionally expressed. From this result, we assume that the gene encoding the aTF will also be transcribed. This does not yet ensure functional translation and folding of the aTF.
- If we do not measure a fluorescence output, mKATE2 is not functionally expressed and it is unlikely that another gene will be functionally expressed under the control of the same promoter and the same RBS. To solve this problem one option would be to exchange the constitutive promoter to ensure functional transcription: Besides Pveg, we also plan to test PliaG and PlepA originating from B. subtilis. Another option is to vary the RBS to ensure functional translation. We plan to use a consensus RBS for B. subtilis from the iGEM parts registry as well as a calculated RBS, obtained from the Salis Lab RBS Calculator.7 Varying the promoter and RBS allows us to identify the most promising combination for the tested parts.
The second step in our test procedure is to investigate whether the inducible promoters PPA1897 and PLux, which were previously used in E. coli, are functional in B. subtilis.1,2 Therefore, we put egfp under the control of the respective promoters and constitutively express the aTFs QscR or LasR. We then measure the green fluorescence output in the presence of 3OC12-HSL. We vary 3OC12-HSL concentrations between 10-4 M and 10-10 M1,2 to assess the sensitivity of the sensing circuit. Besides the inducibility, we also investigate on the question whether 3OC12-HSL passes the Gram-positive cell barrier of B. subtilis. This experimental design was recommended by Prof. Dr. Anke Becker to test if the AHL enters B. subtilis cells.
If we do not measure a green fluorescence output in this experiment, either the aTFs are not functional in B. subtilis or the AHL cannot enter the cell.
An option to find out if the aTFs are functional in B. subtilis is to generate a cell free extract from B. subtilis. With this third experiment, the function of the aTF in the presence of 3OC12-HSL is tested without the cell barrier.8 This can either result in a green fluorescence output or not.
- If we do not measure a fluorescence output, the aTFs are probably not functional in B. subtilis cells.8 This means we would have to adapt our sensing circuit to establish a functional system in B. subtilis.
- If we measure a fluorescence output our sensing circuit is working. Consequently, we then have to prove that the AHL can enter B. subtilis cells.
To test if the AHL can pass through the cell barrier, we need a fourth experimental design. A possible experiment is to add AHL to the medium of a B. subtilis culture and measure the remaining AHL concentration in the medium over time. This measurement could be carried out with analytical methods like mass spectroscopy or gas chromatography. From this experiment, we can expect two outcomes:
- If the AHL concentration in the medium decreases over time, the AHL enters the B. subtilis cells.
- If the concentration of the AHL remains constant, the AHL does not enter the B. subtilis cell. In that case our design for the sensing circuit in B. subtilis would have failed.
Nevertheless, we have vastly researched literature on this topic. Prof. Dr. Anke Becker assumed that AHL molecules should be able to diffuse freely through the cell membrane, as they are small hydrophobic molecules. Additionally, it has been reported that AHL can regulate the gene expression of other Gram-positive bacteria like S. aureus or L. monocytogenes.9,10 This indicates that our fourth experiment likely turns out positive. A more detailed explanation of our research results can be found in the section N-Acyl homoserine lactones and B. subtilis.
After showing functionality of our sensing circuit in B. subtilis, the last steps would be to exchange egfp with recA730 and to test if RecA730 is synthesized in the presence of 3OC12-HSL. Therefore, we would fuse RecA730 with a His6-Tag and detect the expressed protein using anti-His antibodies in a Western Blot. We here expect a band at about 38 kDa6 to confirm the expression of RecA730.1
If this works, we combine the sensing circuit with the genetic switch which shifts the phage from the lysogenic to the lytic cycle. This switch is further explained in the next section.
Bacteriophages
Phage Induction and Release in Non-host Organisms
Our genetic switch is designed to activate phage induction when a quorum sensing molecule from an invading pathogen is detected. We therefore utilize a native mechanism for the induction of lysogenic prophages to lytic phages. For our proof of concept lambda phage induction the production of Cro is necessary. Since cro is regulated by the PR promoter our genetic switch aims to diminish the lambda repressor cI which represses PR. The basic concept of our switch is based on three proteins, where RecA730 and TetR act as antagonists to the lambda repressor cI. The produced TetR binds to the PtetO promoter and herewith prevents production of cI. The produced RecA730 cleaves cI, whereby the PR promoter is no longer repressed and cro is expressed, thus leading to the activation of the lytic cycle.
As proof of concept we want to show that our genetic switch is able to activate the PR promoter by deactivation of cI. Therefore, we used the fluorescent protein eGFP as a reporter under the control of the PR promoter. Also, the genes for recA730 and tetR are regulated by a T7 promoter. Using E. coli BL21 (DE3) the functionality of our genetic switch can then be evaluated through IPTG induction. An overview of our genetic switch can be seen in Figure 3. After a successful proof of concept this genetic switch can be expanded through combination with the genetic circuit for Pathogen Sensing and implemented into Bacillus subtilis.

RecA730 and TetR
Testing of our genetic switch was a great success. We were able to detect a 12-fold increase of eGFP upon induction compared to no induction. See our results here.
Since the controlled production of eGFP worked as expected the next step would be testing the induction of the lysogen. Unfortunately, we were not able to test this in the lab and will develop from here on a theoretical DBTL Cycle.
There are some considerable differences to the native switch in the lysogen. The lambda repressor cI is constitutively expressed in cells containing the lambda prophage, resulting in higher cI concentrations than in our measurements. Eventually, RecA730 activity could be too low to cause efficient deactivation of the repressor resulting in reduced reporter output. This effect could be compensated by either changing regulatory domains for increased RecA730 levels or an knockdown/knockout of the cI gene on the prophage. Necessity of these adaptions could be assessed in identical fluorescence and optical density measurements carried out in the lysogen.
Combination with Pathogen Sensing
If the controlled induction of the lysogen is achieved, the quorum sensing (QS) based switch needs to be integrated into the induction module of our construct. The two proteins RecA730 and TetR are then regulated by the quorum sensing sensitive promoter PPA1897. Also, the allosteric transcription factor QscR needs to be expressed constitutively.
This construct can then be tested with the previously established fluorescence assay. If the quorum sensing controlled degradation of cI is verified again, a similar experiment shall be conducted with the lysogen. This time the optical density should decrease 45 min upon the culture has been exposed to the QS signal.5
Controlled Lytic Cycle Induction and Phage Release in B. subtilis
For the implementation of this system in B. subtilis the phage assembly must be achieved. The highly adapted expression systems evolved in E. coli could be transferred to B. subtilis by variations of regulatory elements and codon optimization. The system responsible for timed cell lysis for the phage release must also be adapted for the new host organism. This could be achieved by protein engineering of holines, membrane pores for cell lysis. Alternatively, transcription and translation of these holines could be tuned yielding in the desired delay between pathogen sensing and lysis. For integration of the phages, we are proposing a phage integration site similar to systems inducing the lysogenic state in lambda phages.
To adapt our system to specifically lyse pathogens, the genetic switch must be adapted to regulatory systems of respective bacteriophages targeting the desired pathogens. The implementation of the lambda regulatory system displays a proof of concept for the inclusion of lysogenic-lytic regulatory systems of prophages. This highlights the feasibility to extend a similar approach to multiple lysogenic phages, matching our idea of a modular approach adaptable to a multitude of phages.
We do recognize there are still many major challenges to achieve the complete synthesis and assembly of functional phages in a non-host organism. However, we think these are helpful and necessary steps to overcome the remaining hurdles through an iterative approach in the form of a DBTL cycle.
Biosafety
Inversion via Cre-Lox Recombination
As we further developed the kill-switch design of last year’s iGEM Team of TU Darmstadt, we generally optimized the schematic framework by adding RBS and terminators to enable transcription and translation. As previously mentioned in Adaptation of Parts, we considered required changes within our construct to adapt to E. coli as new chassis. Thus, the kill-switch design was fitting to conduct potential assays in B. subtilis as well as E. coli.
In case of our proof of concept assay turns out to be successful in E. coli, we can assume that, due to the conserved lox sites, the inversion of the final cassette should be functional in B. subtilis as well. Nevertheless, we are aware that information and findings gathered in E. coli cannot be directly transferred to B. subtilis since we switch between two different organisms which differ in several aspects, e.g., the membrane architecture [from Gram-negative E. coli to Gram-positive B. subtilis). Additionally, E. coli is lacking the two-component ComQXPA-system.11 Hence, we need an extended proof-of-concept assay to test whether the ComX signaling is functional in B. subtilis. This could be achieved in a DBTL process based on the framework of our current genetic circuit for B. subtilis. If meeting this requirement as well, the final kill-switch cassette would be eventually implemented in the biofilm forming strain DK1042. Functional ComX can be heterologously produced in E. coli.12,13 Recombinant ComX can then be added externally to the system to induce expression of the essential gene rpsB. As mentioned in the previous section Theoretical Elaboration: B. subtilis, the final cassette design (Assay 4) contains a bidirectional promoter cassette flanked by lox66 and lox71 with constitutive Pveg and QS-inducible PdegQ promoter. After Cre-Lox inversion of the cassette, expression of the essential gene should be induced by the transcription factor ComA-P.14
Since the functionality of our final kill-switch fully relies on the ComQXPA-system, we are aware of the fact that our engineered kill-switch cassette up to this point might not be optimal for implementation in our biofilm. To address potential weaknesses several parameters can be adjusted to yield an optimized kill-switch.
We could face the issue that the cells are not viable under transcriptional control of PdegQ. Cell death can be detected via Live/Dead staining. Since PdegQ is a non-native promoter for the rpsB gene, the transcription efficiency might be too low to achieve a sufficient expression level of rpsB. In this case, we could try to optimize the binding affinity of the RNA-polymerase by modulating the operator region upstream of the promoter core motif of PdegQ.15
On the translation level, we could take RBS shuffling into account. Thus, we would be able to adapt spacing between the core Shine-Dalgarno motif and either start codon or PdegQ promoter16, while not affecting the –35 and –10 regions. This way, we would try to improve translational efficiency.
Adding a N-terminal His-tag to recombinant proteins can improve expression levels.17,18 In this case, we would test if rpsB further maintains proper function with addition of a N-terminal His-tag.
Furthermore, it would also be an option to replace the essential gene. Doing this, we would still focus on translational control since replication is not constantly needed and thus not that critical for the survival of the cell in the first place. Choosing the essential gene is challenging, as it is way easier to control a gene separately than within an operon containing several regulatory genes.
A potential alternative would be thrS coding for the threonyl-tRNA synthetase, being part of a regulon. But its paralogue thrZ must be knocked out additionally for cell viability control.19
Given the fact that the kill-switch is completely functional within sporulation-deficient B. subtilis 16820, we could further optimize the chassis and try to implement our system in a B. subtilis strain with a minimal genome.21,22 Doing this, we would have to make sure that essential genes for biofilm formation are not excluded. Thus, we could minimize potential interactions of our kill-switch system and make it more independent from environmental conditions, thereby positively affecting biocontainment efficiency.
All in all, empirical optimization, including multiple DBTL-iterations, will be required to engineer the optimal kill-switch, thus ensuring biocontainment of our system for application outside of the lab. This way, our genetic construct could provide a reliable system to prevent the uncontrolled release of GMOs easily adaptable to a multitude of biofilm based systems.
References
- 1. Wu Y, Wang C-W, Wang D, Wei N. A Whole-Cell Biosensor for Point-of-Care Detection of Waterborne Bacterial Pathogens. ACS Synthetic Biology. 2021:333–344. http://dx.doi.org/10.1021/acssynbio.0c00491. doi:10.1021/acssynbio.0c00491
- 2. Saeidi N, Wong CK, Lo T, Nguyen HX, Ling H, Leong SSJ, Poh CL, Chang MW. Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Molecular Systems Biology. 2011:521. http://dx.doi.org/10.1038/msb.2011.55. doi:10.1038/msb.2011.55
- 3. Venturi V. Regulation of quorum sensing inPseudomonas. FEMS Microbiology Reviews. 2006:274–291. http://dx.doi.org/10.1111/j.1574-6976.2005.00012.x. doi:10.1111/j.1574-6976.2005.00012.x
- 4. Lequette Y, Lee J-H, Ledgham F, Lazdunski A, Greenberg EP. A Distinct QscR Regulon in the Pseudomonas aeruginosa Quorum-Sensing Circuit. Journal of Bacteriology. 2006:3365–3370. http://dx.doi.org/10.1128/JB.188.9.3365-3370.2006. doi:10.1128/jb.188.9.3365-3370.2006
- 5. Chapter One: The master elements of control. In: A Genetic Switch: Phage Lambda Revisited. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2004.
- 6. Vlašić I, Šimatović A, Brčić-Kostić K. Genetic Requirements for High Constitutive SOS Expression in recA730 Mutants of Escherichia coli. Journal of Bacteriology. 2011:4643–4651. http://dx.doi.org/10.1128/jb.00368-11. doi:10.1128/jb.00368-11
- 7. RBS Calculator. Design Ribosome Binding Site Sequences to Control Protein Expression Levels. De Novo DNA. 2021 [accessed 2021 Jul 7]. design_rbs_calculator
- 8. Kelwick R, Webb AJ, MacDonald JT, Freemont PS. Development of a Bacillus subtilis cell-free transcription-translation system for prototyping regulatory elements. Metabolic Engineering. 2016:370–381. http://dx.doi.org/10.1016/j.ymben.2016.09.008. doi:10.1016/j.ymben.2016.09.008
- 9. Qazi S, Middleton B, Muharram SH, Cockayne A, Hill P, O’Shea P, Chhabra SR, Cámara M, Williams P. N-Acylhomoserine Lactones Antagonize Virulence Gene Expression and Quorum Sensing in Staphylococcus aureus. Infection and Immunity. 2006:910–919. http://dx.doi.org/10.1128/iai.74.2.910-919.2006. doi:10.1128/iai.74.2.910-919.2006
- 10. Naik MM, Bhangui P, Bhat C. The first report on Listeria monocytogenes producing siderophores and responds positively to N-acyl homoserine lactone (AHL) molecules by enhanced biofilm formation. Archives of Microbiology. 2017:1409–1415. http://dx.doi.org/10.1007/s00203-017-1416-8. doi:10.1007/s00203-017-1416-8
- 11. Dogsa I, Choudhary KS, Marsetic Z, Hudaiberdiev S, Vera R, Pongor S, Mandic-Mulec I. ComQXPA quorum sensing systems may not be unique to Bacillus subtilis: a census in prokaryotic genomes. PloS one. 2014;9:e96122. doi:10.1371/journal.pone.0096122
- 12. Tortosa P, Logsdon L, Kraigher B, Itoh Y, Mandic-Mulec I, Dubnau D. Specificity and genetic polymorphism of the Bacillus competence quorum-sensing system. Journal of bacteriology. 2001;183:451–460. doi:10.1128/JB.183.2.451-460.2001
- 13. Ansaldi M, Marolt D, Stebe T, Mandic-Mulec I, Dubnau D. Specific activation of the Bacillus quorum-sensing systems by isoprenylated pheromone variants. Molecular microbiology. 2002;44:1561–1573. doi:10.1046/j.1365-2958.2002.02977.x
- 14. Roggiani M, Dubnau D. ComA, a phosphorylated response regulator protein of Bacillus subtilis, binds to the promoter region of srfA. Journal of bacteriology. 1993;175:3182–3187. doi:10.1128/jb.175.10.3182-3187.1993
- 15. Stanley NR, Lazazzera BA. Defining the genetic differences between wild and domestic strains of Bacillus subtilis that affect poly-gamma-dl-glutamic acid production and biofilm formation. Molecular microbiology. 2005;57:1143–1158. doi:10.1111/j.1365-2958.2005.04746.x.
- 16. Tietze L, Lale R. Importance of the 5’ regulatory region to bacterial synthetic biology applications. Microbial biotechnology. 2021. doi:10.1111/1751-7915.13868
- 17. Park W-J, You S-H, Choi H-A, Chu Y-J, Kim G-J. Over-expression of recombinant proteins with N-terminal His-tag via subcellular uneven distribution in Escherichia coli. Acta biochimica et biophysica Sinica. 2015;47:661. doi:10.1093/abbs/gmv069
- 18. Shilling PJ, Mirzadeh K, Cumming AJ, Widesheim M, Köck Z, Daley DO. Improved designs for pET expression plasmids increase protein production yield in Escherichia coli. Communications biology. 2020;3:214. doi:10.1038/s42003-020-0939-8
- 19. Reu DR, Commichau FM, Gundlach J, Zhu B, Stülke J. The Blueprint of a Minimal Cell: MiniBacillus. Microbiology and molecular biology reviews : MMBR. 2016;80:955–987. doi:10.1128/MMBR.00029-16
- 20. Špacapan M, Danevčič T, Štefanic P, Porter M, Stanley-Wall NR, Mandic-Mulec I. The ComX Quorum Sensing Peptide of Bacillus subtilis Affects Biofilm Formation Negatively and Sporulation Positively. Microorganisms. 2020;8. doi:10.3390/microorganisms8081131
- 21. Sung BH, Choe D, Kim SC, Cho B-K. Construction of a minimal genome as a chassis for synthetic biology. Essays in biochemistry. 2016;60:337–346. doi:10.1042/EBC20160024
- 22. Michalik S, Reder A, Richts B, Fahauer P, Mäder U, Pedreira T, Poehlein A, van Heel AJ, van Tilburg AY, Altenbuchner J, et al. The Bacillus subtilis Minimal Genome Compendium. ACS synthetic biology. 2021. doi:10.1021/acssynbio.1c00339