
Introduction
A key aspect of this year’s project PHIRE BYRD – Phage mediated Immune Response by Recognizing, Defensive sleeper cell – was the creation of a biofilm that contains multiple sleeper cells. A sleeper cell is a B. subtilis cell that remains in a dormant “sleeper cell” state until being activated. This biofilm is not only supposed to contain one kind of B. subtilis sleeper cell but multiple differently genetically modified B. subtilis sleeper cells from the strain DK1042.1 Since the genetic uptake of B. subtilis is limited, one sleeper cell will carry the genome of one bacteriophage that targets a specific pathogen. This way our emerging multi-functional B. subtilis biofilm will then be a robust phage-defense system against various pathogens (Figure 1).
The combination of different phages leads to a modular defense system that is effective against more than just one pathogen, e.g., Pseudomonas aeruginosa, and results in a powerful protection of our biofilm against any kind of pathogen. Additionally, our created biofilm should be homogenous in order to achieve uniform protection. See our lab results for further information.

Before new, improved biological systems such as functionalized biofilms can be released, it must be ensured that they do not cause new problems. The use of biofilms in wastewater treatment, the protection of plant roots from parasites, or the synthesis of biochemical products carries the risk of pathogens, like Pseudomonas aeruginosa, implanting themselves into the biofilm.
We therefore want to design a system that makes the use of functionalized biofilms risk-free by equipping the biofilm with a self-defense mechanism!
B. subtilis and Biofilm Theory
Bacillus subtilis
For the phage-defense system including functionalized biofilm we selected Bacillus subtilis as our biofilm forming organism.
B. subtilis is a Gram-positive bacterium that can usually be found in soil. Like all Gram-positive bacteria, it features only one cell membrane and is therefore often used for protein secretion.2 Furthermore, B. subtilis is characterized as a facultative anaerobic organism.3,4 This means that it can grow in oxygenic as well as in anaerobic conditions. This can be an advantage if the phage-defense system is used to protect functionalized biofilms for water treatment3 or the protection of plant roots from parasites.
In its natural habitat B. subtilis naturally forms biofilms on plant roots as it needs them as a nutrient source. This has made it a popular model organism for studying biofilms.5
By using a natural biofilm former, it is easier for our team to predict and assess biofilm formation as we do not have to consider how and under which particular circumstances the organism will form the desired biofilm. Another advantage of using B. subtilis is that is classified “generally recognized as safe” short GRAS by the American Food and Drug Administration (FDA). This is a very important aspect for us, given that we want to protect functionalized biofilms in wastewater treatment plants or the agriculture.6
DK1042
Bacillus subtilis is not only a popular model organism for studying biofilms3 but also a popular genetic model systems due to its fast growth as dispersed cells and its capacity to absorb and include exogenous DNA through herbal competence.1 The B. subtilis strain NCIB3610 retained many of its natural biological characteristics that have been removed from domesticated strains such as 168 (e.g., formation of the air-liquid interface biofilms called pellicle biofilms).1 Unfortunately, this strain is not competent enough for unproblematically genetic modifications1 and its natural competence cannot simply be induced.7 By improving NCIB3610’s 84-kb endogenous plasmid, called pBS32, it is possible to increase its eligibility for genetic modifications.1 This is possible because of the protein ComI, which is encoded by the endogenous plasmid, that is interfering with the competence machinery of NCIB3610.1
The strain we use for our proof of concept DK1042 is the ComIQ12L mutant of NCIB3610.1 It is therefore genetically modifiable and carries the natural biological characteristics of B. subtilis like the ability to form biofilms.
Biofilm and Biofilm formation
A bacterial biofilm is a community consisting of a single or multiple bacteria species that produces an extracellular matrix.8
This biofilm matrix is made of several components including exopolysaccharides, proteins, lipids and extracellular DNA. These components are produced by the inhabiting bacteria.9 It varies in its composition from species to species. Some of the structural elements of the matrix can also possess special characteristics that are beneficial to the biofilm i.e., B. subtilis uses proteins called hydrophobins to form highly hydrophobic biofilms that float at the air-liquid interface.8 But notwithstanding the above, the biofilm matrix provides crucial support for the structure and organization of any kind of biofilm, as well as protection for the microbial communities living in them.
Various natural and synthetic laboratory conditions lead to the formation of different types of biofilms. Several have been used to study the microbial biofilm formation of B. subtilis. The different types include colonial biofilms at the air-agar interface and liquid biofilms at the air-liquid interface (the kind of biofilm we are cultivating), also known as pellicle. In the case of certain strains, which have been domesticated there is a third type of biofilm: immersed surface-adherent biofilms at the liquid-solid junction.5

In case of a classical biofilm the bacterial cells that are situated in the liquid phase settle on top of a surface like a root or a pipe. In general, hydrophobic and coarse surfaces are preferred over hydrophilic and smooth surfaces, because biofilm forming microorganisms can attach more firmly to these types of surfaces. The reason for the enhanced attachment is that the hydrophobic connections will in general increment with an expanding nonpolar nature of either the microbial cell surface or the surface the bacteria settle on.11 Once the adhesion between the cells and the surface is sufficient, the bacteria use the available nutrients to reproduce rapidly. After this rapid growth phase, the adhesion of the microorganisms to the surface is extremely strong.10
The maturation of the emerged biofilm leads to cell-cell communication between the microbial cells located in it. This communication takes place via the secretion of autoinducer signaling molecules. B. subtilis cells therefore secrete peptides with various submit-translational changes that can be recognized by other B. subtilis cells both by a membrane-sure histidine kinase or cytoplasmic receptors.12 Moving further along in the maturation process the bacterial cells are divided into several communities, each of them responsible for a particular job.13 Both developments in the maturation process are accompanied by the expansion of the cell population living in the biofilm and therefore also by an expansion of the biofilm itself. During the expansion process various complex diffusion pathways are being created that are used for the distribution of nutrients, oxygen and other elements which are needed for the growth and development of cells.14
When these bacterial cells accumulate, and the signaling molecule concentration reaches a sufficient level, the final stage of the biofilm formation process is induced: the dispersion of matured biofilm cells and fragments. It is characterized by the detachment of the biofilm, and the return of the sedentary cells to their previous mobile and unattached form. This way, the biofilm is able to spread, settling in new locations where it can form colonies and new biofilms.8
Functions of a Biofilm
Biofilms feature characteristics that we believe are ideal for the implementation of our sleeper cell defense system.
In order to enable our sleeper cells to defend a biological system against several pathogens at the same time, like Pseudomonas aeruginosa, they need to be successfully integrated into a living biological system. Since biofilms naturally show a high degree of biodiversity, featuring different bacteria strains and species15, it is possible to integrate a sleeper cell into a biofilm or to find a biofilm forming bacteria strain whose cells can be transformed to form a sleeper cell, which protects its biofilm.
The microorganisms within such a biofilm tend to have a high degree of cell-cell contacts, as well as a very low mobility compared to planktonic cells, which results in a biofilm containing a significant number of organisms per unit volume.8 This dense living community is surrounded and stabilized by the so-called matrix. It mainly consists of extracellular polymeric substances (EPS) and creates a compartment which is more or less isolated from the environment. This means that the microorganisms mainly reside in the biofilm, partly because they are protected from toxic substances and harsh environmental conditions, such as extreme temperatures or extreme pH-levels.9
Thus, working with a biofilm means utilizing a system that has increased stability and resistance without any additional modifications and an increased level of localizability. Conversely, this means that such a compact compartment is ideal for the introduction of genetical modifications in microorganisms since combining it with a kill-switch, as an extra safety precaution, ensures that these GMOs are not likely to survive outside the biofilm.

In addition to the already mentioned properties, biofilms also possess other interesting characteristics that may be relevant for the field of implementing improved biological systems like functionalized biofilms. Biofilms are increasingly being used. Especially in the development of environmentally friendly options for existing processes. To list a few examples from former iGEM Teams: Groningen used the ability of biofilms to host multiple species to develop a root protecting patch in 2020.16 Also, the iGEM Team Darmstadt 2020 developed a biofilm that filters out diclofenac from wastewater, by engineering the TasA structure of the biofilm and expressing laccases on it.17
Multi-functional biofilm
The overall aim of our project is not only to create a biofilm that detects and kills Pseudomonas aeruginosa, but to create a modular system to do so. Our project goal is to introduce various bacteriophage genomes into Bacillus subtilis host cells. Thus, we aim to create B. subtilis strains of which each is able to produce a different phage. A biofilm containing a variety of these differently modified B. subtilis cells should possess a broad defense spectrum against multiple pathogens. The composition of the cells in the biofilm could be adjusted depending on the application and the expected pathogens in this application. A schematic overview of such a biofilm can be seen in Figure 4.

To create a multi-strain biofilm, a stable multi-strain co-culture is required beforehand. Extensive research showed different methods for creating and ensuring a resilient and stable co-culture. These methods include the generation of metabolic dependency and the usage of quorum sensing or surface proteins. Furthermore, to ensure that the biofilm is evenly protected, the B. subtilis strains should be evenly distributed within the co-culture biofilm.
After discussing our plans with experts on B. subtilis and biofilms, we concluded that closely related B. subtilis strains should form a stable co-culture without the introduction of engineered interdependencies. None of the strains should be favored with regards to selection pressure, and they should not interfere with each other in in a way that would be disadvantageous for our application. Thus, it would be possible to simply mix different strains in the same ratio with each other. Concerning our question whether the biofilm is evenly distributed, Prof. Daniel Kearns suggested that we should focus on using only a single strain that is labelled with different fluorescence proteins, as this would greatly reduce assay complexity. Therefore, from now on we speak of a multi-functional biofilm, not multi-strain.
In the case that we observe that co-cultures of different B. subtilis strains are either unstable or unevenly distributed, we can revert to three established methods to create stable co-cultures. An overview of these methods is given below.
Since our overall project aim is to create a self-defensive biofilm, it is important that the co-culture possesses the ability to form biofilms. We want to analyse the formed biofilm in regard to different aspects such as the distribution of the strains, as well as the overall stability. A more detailed explanation of our planned procedures in the lab, as well as the actual implementation of such can be found below.
Methods for stability and homogenity
Quorum Sensing
An approach to tackling the potential homogeneity and stability problem of our co-culture was the use of a quorum sensing (QS) based control mechanism. It has been shown recently that the engineering of a QS-based system influencing the co-culture composition in E. coli is possible. Although the mentioned system uses external regulation of the signaling molecule Autoinducer-2 (AI-2), the introduction of AI-2 secretion capabilities into the used strains is, according to the articles authors, possible.18
As our goal is the creation of a same species multi-functional biofilm, the described use of a species-specific inhibitor would not have been feasible. Furthermore, the proposed use of AI-2 as a marker could have introduced problems downstream, as AI-2 takes a key role in the colony organization and biofilm formation in B. subtilis.19,20
Although B. subtilis features a well-developed QS-system that is used for cell-cell communication21,22, we did not find literature showing that engineered control of B. subtilis growth through use of QS-systems had been implemented before. Even though it is not of importance for our approaches at this point, this might be a promising concept for future research.
Due to our experts already telling us that, in their opinion, a system to control the homogeneity and stability of our co-culture should not be necessary, and the fact that we had two different approaches (metabolic dependency and cell-cell adhesion), which seemed to have more potential, we decided that further research into this topic would be out-of-scope for our project.
Metabolic Dependency
During the search for methods to ensure stable co-culture growth, we came across the field of synthetically created microbial communities. Currently, research in this area is mainly conducted on microbial communities featuring E. coli, but the results on these experiments look promising.23–25 Although we did not find a source that confirmed that the creation of a stable co-culture in B. subtilis through metabolic dependency was possible, we were intrigued by the findings in E. coli.
We tried to find sources that documented if such behavior had been observed naturally in B. subtilis, as this would mean that our idea could, at least in theory, be feasible. The work of Liu et al. 2015 indeed showed a natural occurrence of metabolite cross-feeding in the process of biofilm formation in B. subtilis.26 Although this interaction was in general not mandatory for the survival of the bacteria, it nevertheless meant that such interactions are possible and indeed a way for co-culture regulation. In our talk with Dr. Bischofs, she mentioned that there had been research on auxotrophic B. subtilis cultures in the past, which could help us in our endeavor to engineer a synthetic co-culture. However, in her experience such a method should not be needed for stable co-culture creation.
After receiving this information, we found two papers focusing on the survivability of auxotrophic B. subtilis strains through cross-feeding interactions.27,28
If the degree of homogeneity of our co-culture or its stability would have, against our experts’ expectations, become a problem, we could have tried to use the information we gathered to engineer two differently auxotrophic strains. By doing so, we would create a mandatory cross-feeding relationship between the strains, forcing a homogenous and stable co-culture.
Bacterial Cell-Cell Adhesion
One possible approach to achieve a desired level of synthetic homogeneity is to couple two or more strains via heterologously expressed proteins at the cell surface. This idea has already been implemented with E. coli.29
The basis of this method is a construct which is expressed on the surface of the cell. This construct faces the outside carrying an antibody or an antigen. If two organisms carry matching constructs, they can interact with each other via adhesion. To understand the method, one must deal with the basic structure, which consists of a non-adhesive part.
The non-adhesive part of the construct is a shortened version of Intimin and consists, in the correct order of an export tag, a LysM domain binding to peptidoglycan, a ß-barrel and a spacer to which the adhesin is then attached to.29 The expression of the construct regulated by a TetR or AraC repressor. Intimin, also known as autotransporter-III and thus a component of the type III secretion system, is a membrane-exciting protein found in the strains Yersinia spp., pathogenic E. coli and Citrobacter spp.30 A ß-barrel is a membrane-bound protein that has a barrel-like structure and is found only in the outer membrane of chloroplasts, mitochondria and Gram-negative bacteria.31 Since B. subtilis is a Gram-positive bacterium it does not possess Intimin and its secretion system, as well as a ß-barrel, consequently an alternative approach to protein display must be found for the implementation of the idea.
The expression and presentation of heterologous proteins can be approached either in a covalent or non-covalent way. A non-covalent method has already been presented by Chen et al.32 There, the protein was fused with LysM. With this method up to an estimated 1.1 x 108 units could be displayed with ß-lactamase. A covalent method is demonstrated by Spirig et al.33 There, a protein that has an LPXTG sorting signal and the enzyme sortase A attached to its C-terminus is connected to lipid II molecules by the aforementioned enzyme and thus ultimately integrated into the naturally occurring peptidoglycan structure. Whether a non-covalent or a covalent method is more suitable for the biofilm needs to be experimentally determined. In most cases, non-covalent approaches are easier to implement, but covalent methods usually offer more reliability and resilience.
Now that the basic structure is known, it is still important to deal with the functional part of the construct, which is the adhesive part, consisting of an antibody or a matching antigen. The antibody is a camelid antibody which can be expressed and presented on the surface. Single domain camelid antibodies are advantageous due to their small size of about 125 amino acids and their increased tolerance to various environmental conditions as well as their lower number of disulphide bridges.34
Starting with a pre-made construct and a useful display method, a library of strains with different adhesins can now be generated as demonstrated by Glass et al.29 This library should be orthogonal to avoid side effects (i.e., that adhesins only interact with designed partners) and composable to be able to link multiple strains (i.e., that arbitrary combinations of multiple adhesins function simultaneously within one cell). Such a library can be generated by screening all available adhesins pairwise interactions via precipitation assays.
With the resulting library, one has the option to control the adhesion strength between cells quantitatively, as well as the morphology. To control the adhesion strength and thus also the avoidable mobility, there are different approaches:
- control of the expression of the construct and thus the number of binding constrictions by the concentration of the inhibitor of the repressor29;
- control of the binding strength of the adhesins by selecting weak or strong affinity antibody-antigen pairs29;
- reducing adhesin interactions by adding a competitively binding protein.29
In addition to the total homogeneity we are aiming for, this approach also includes the ability to form a variety of complex patterns. Some of the patterns might turn out to be more advantageous for our cause depending on the cultures that are supposed to be protected.
- Differential adhesion, for example, could provide maximum protection to a biofilm if the sleeper cells are in the periphery, as pathogens primarily attack the biofilm there.
- Phase separation on the other hand offers an interesting contrast to the desired goal of homogeneity. If it turns out that homogeneity is not needed or even not desired, a multi-strain biofilm that would naturally form a homogeneous biofilm can be separated in this way.
- The coaggregation bridging method shows that more than just two strains can be brought into a structure.
Although it has been shown that adhesion does not stand in the way of cell guarding29, the core of this method is a shape-giving one. The stronger the interaction, the more consolidated the positions and vice versa. For a consolidated structure, it is important to investigate to what extent it is maintained over several generations or whether local conglomerates form and what effects the resulting reduced mobility has. In the case of very loose structures, maintenance of the intended structure over the generations must be examined. Alternatively, the largely preserved motility could allow for example separation of the tribes.
Theoretical Lab Plan
The overall aim of this part of the project was to create a multi-functional Bacillus subtilis biofilm to analyse its characteristics and to assess the question whether such a biofilm containing several phage genomes in co-culture would be stable and homogeneously distributed.
A possible application of our self-defensive biofilm is in wastewater treatment. We consequently chose to grow pellicle biofilms, which form at the air/liquid interface.
The experts we discussed our project with pointed out that there is no real quantitative assay to determine the quality of a B. subtilis biofilm. To determine the quality of a formed biofilm a few macroscopically visible parameters, e.g., the surface texture and the overall look of the biofilm, can be evaluated, mainly based on practical experiences. In the case of co-cultured biofilms, formed biofilms are compared to the biofilms formed by the respective monocultures. This could reveal obvious biofilm stability issues of the multi-functional biofilms, justifying further investigation into the before mentioned methods for stability improvement.
In order to offer a uniform protection throughout the whole biofilm, it also requires a homogeneous distribution of the different B. subtilis cells on a microscopic level. To assess the distribution of cells in the biofilm, we decided to integrate expression cassettes of fluorescent proteins into the genome of B. subtilis. Each fluorescent protein represents a respective phage genome. This allows us to determine the overall distribution by quantitative methods like distribution analysis in the frequency domain, as described by Prof. Dr. Ákos Kovács and co-workers.35

FlowChamber
As we are working with biofilms, a major concern was not only the stability of our co-culture, but also the physical stability of our multi-functional biofilm. That is why we decided to reiterate on the FlowChamber design from the TU Darmstadt 2020 team.
After consulting with members of the 2020 TU Darmstadt iGEM team, the numerous flaws in the old design became apparent. Only fixing the flaws wouldn’t be that effective, therefore we decided that a complete redesign incorporating the feedback from last year would be more useful and promising.
One major problem with the chamber of 2020 was the entrance and exit of flow perpendicular to the sample. This resulted in a disruption of flow and consequently the formation of vortices, potentially trapping separated pieces of biofilm in the chamber, affecting the results.
In order to improve and prove the capabilities of the new FlowChamber we designed the new iteration in a CAD program (Figure 6).
The most important improvements are a gentle diversion of flow, which enters the chamber parallel to the sample surface, using a parabola shaped deflector incorporated into the top part, and the introduction of a seal into the design.
In order to simulate flow through the chamber before construction we decided to conduct a computational fluid dynamics (CFD) simulation, which numerical solves a set of second order partial differential equations to predict the behavior of a fluid under the chosen conditions.36 These simulations can include the simulation of friction and/or the simulation of vortex formation. In our case we are only interested in an approximate model of the flow through the chamber, helping us to design an area that exhibits suction forces onto the sample, as well as designing a flow that flows as freely as possible, preventing large amounts of sample being recycled in the chamber instead of reaching our sample that we measure later on.

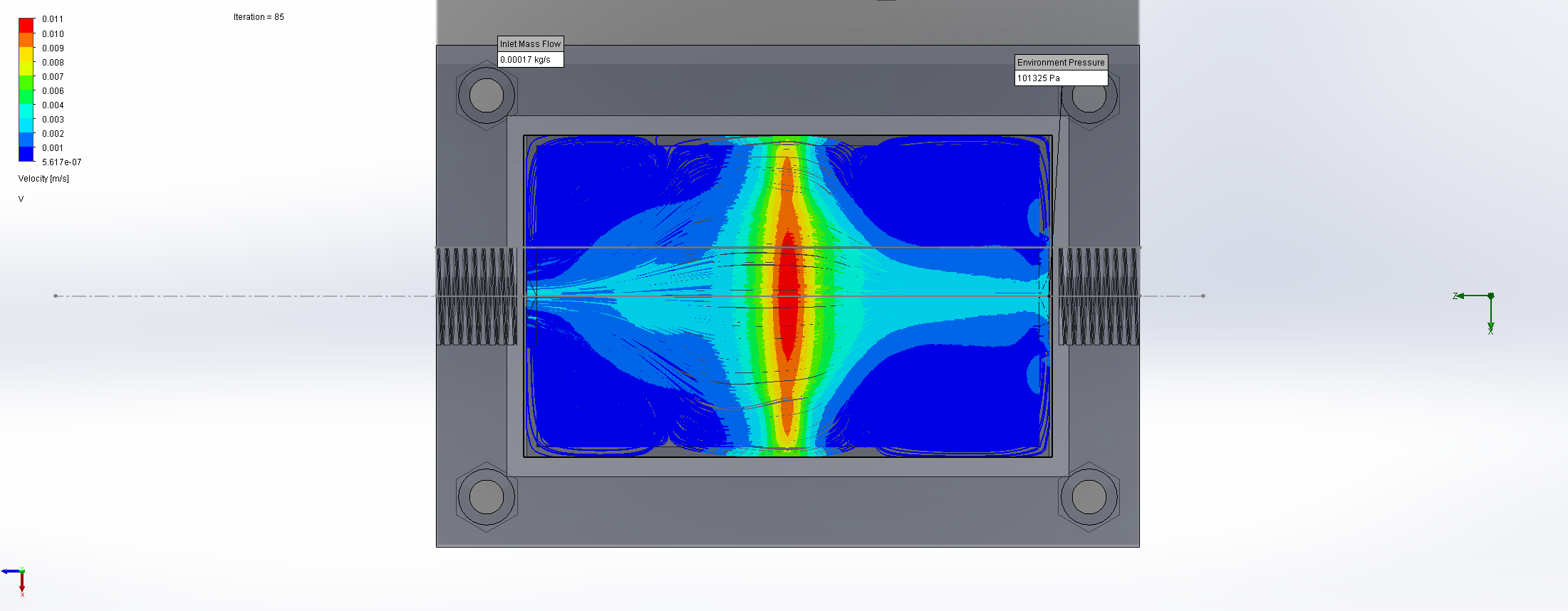
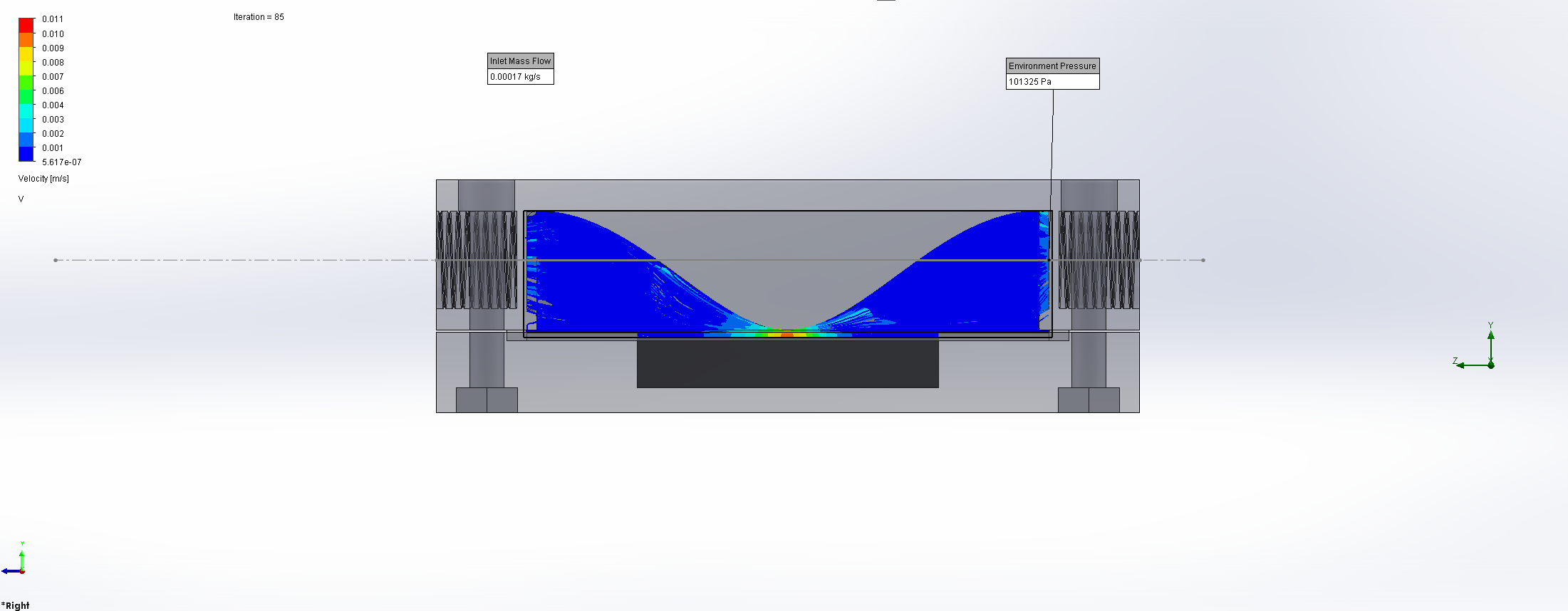
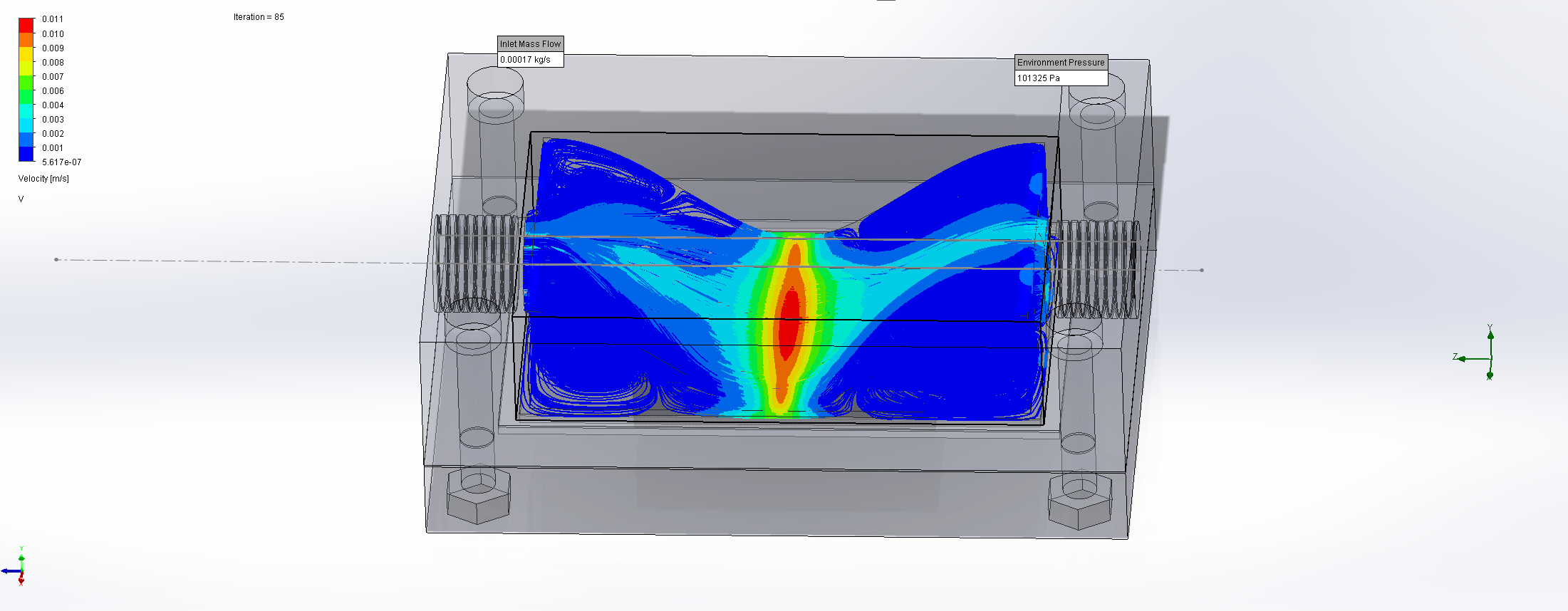
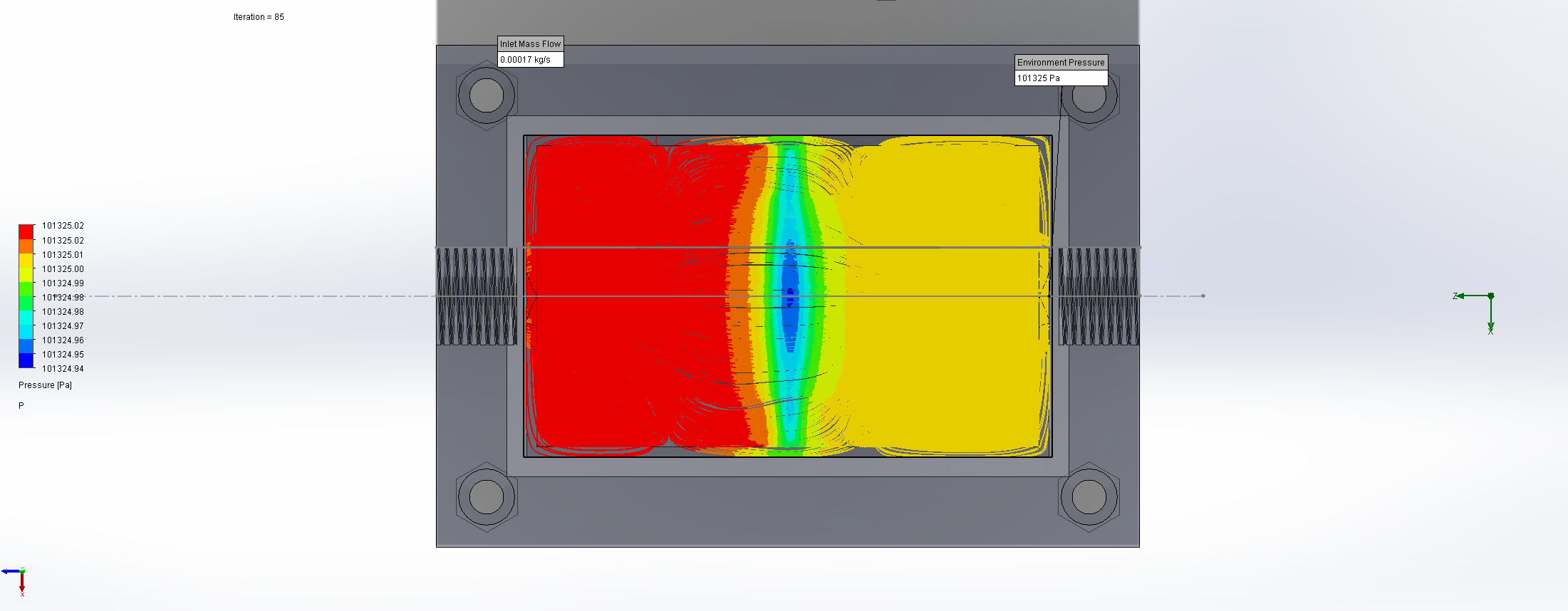
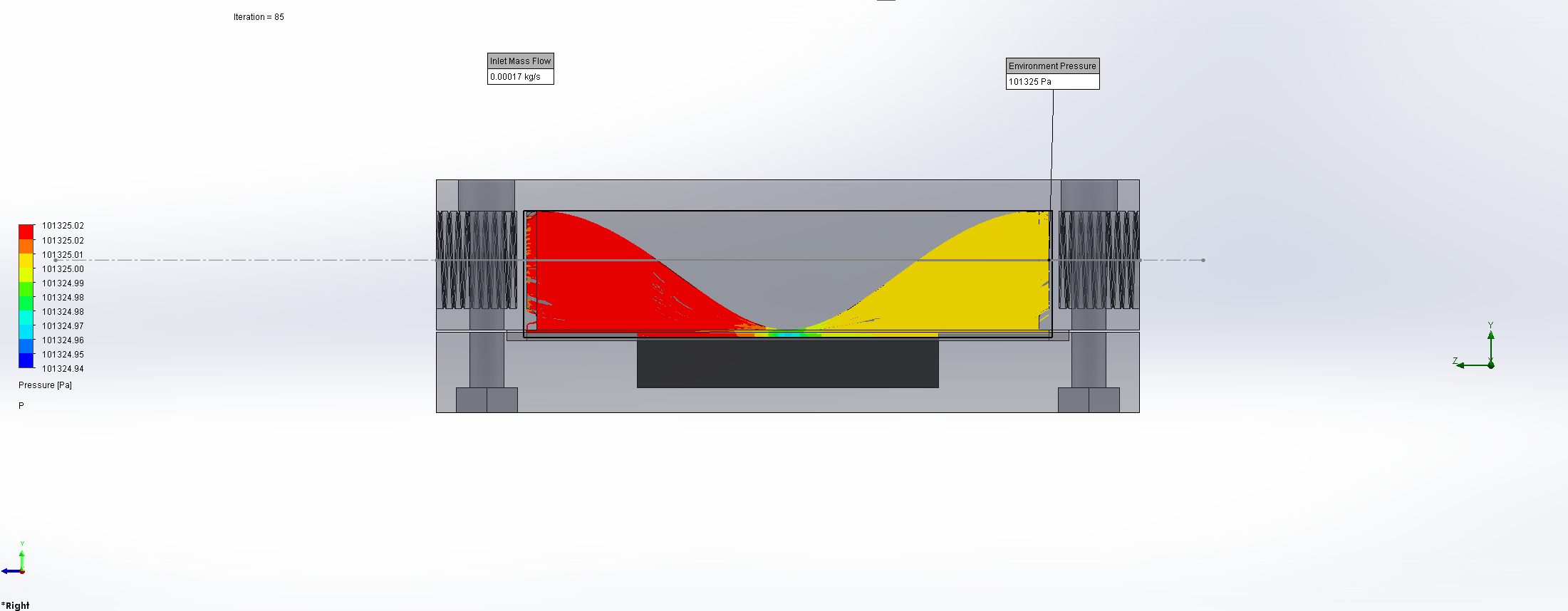
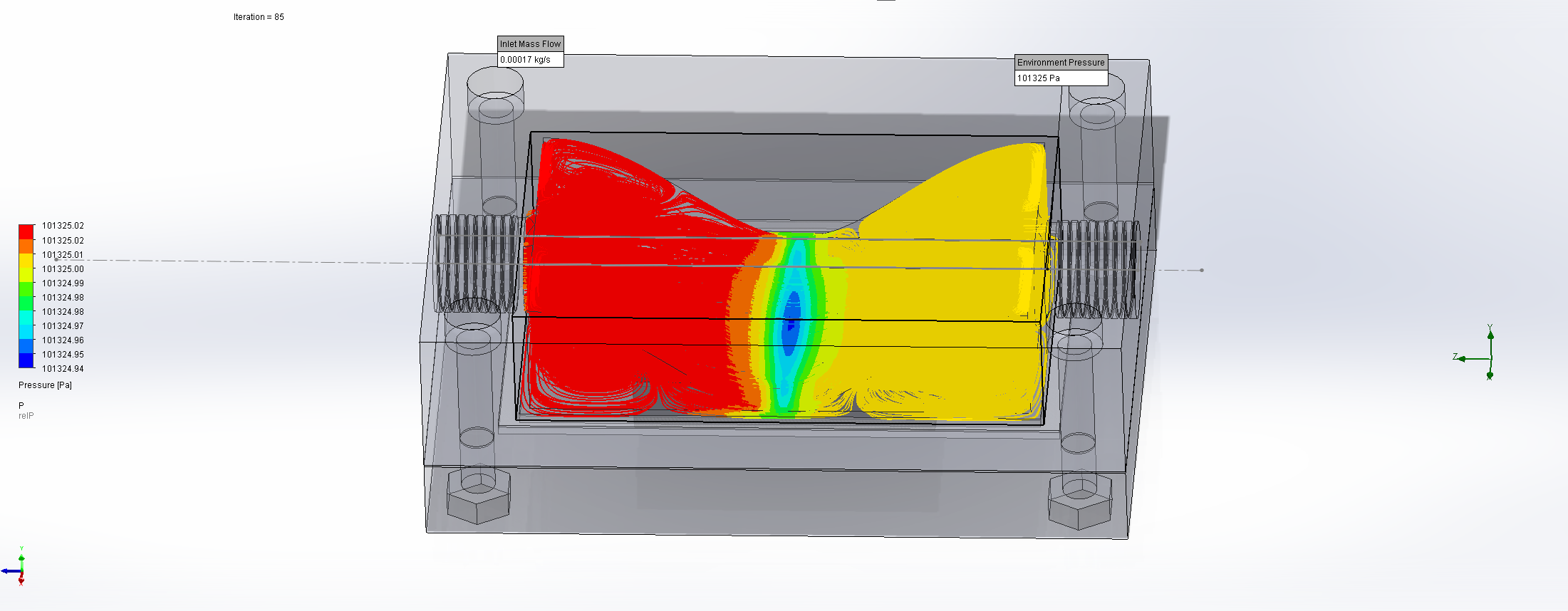
For this we utilized the FlowAnalysis Module of SolidWorks to conduct an internal flow simulation, showing the differences in pressure and velocity as well as vortices formation, which could influence the accuracy of our results. The results of our FlowAnalysis can be seen in the Figures 7, 8 and 9.

Top 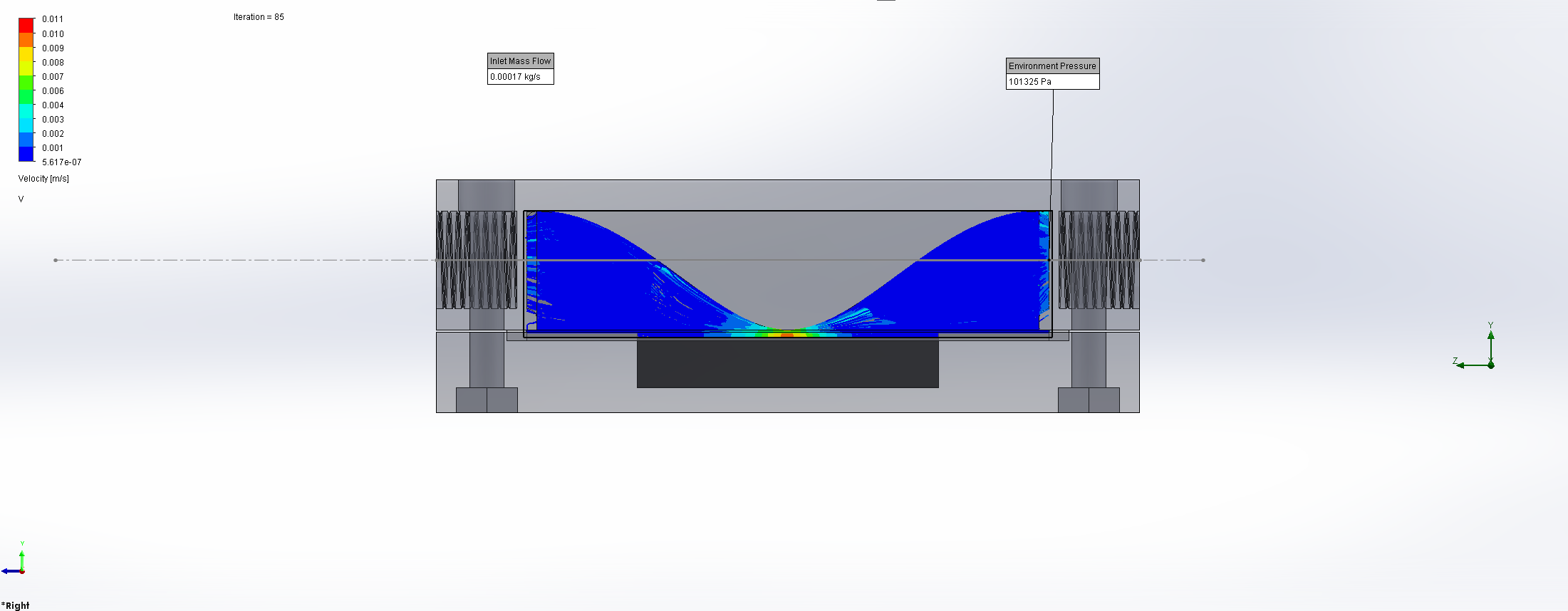
Right 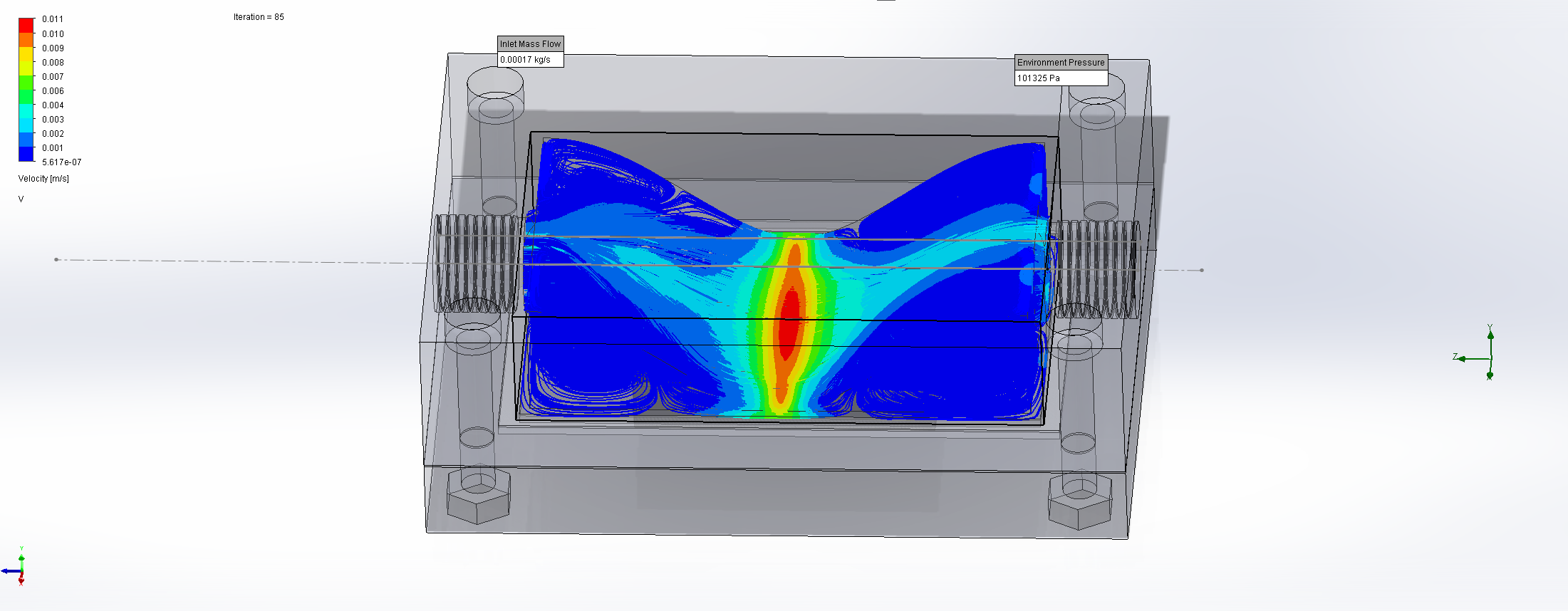
Isometric

Top 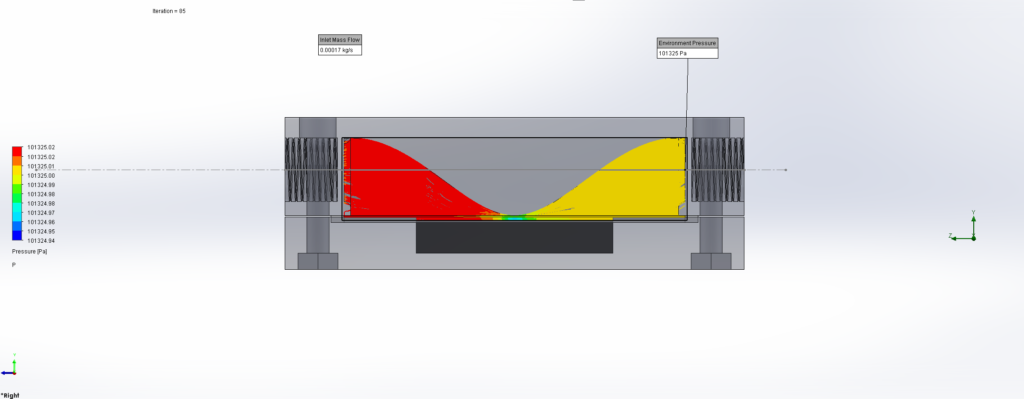
Right 
Isometric

Top 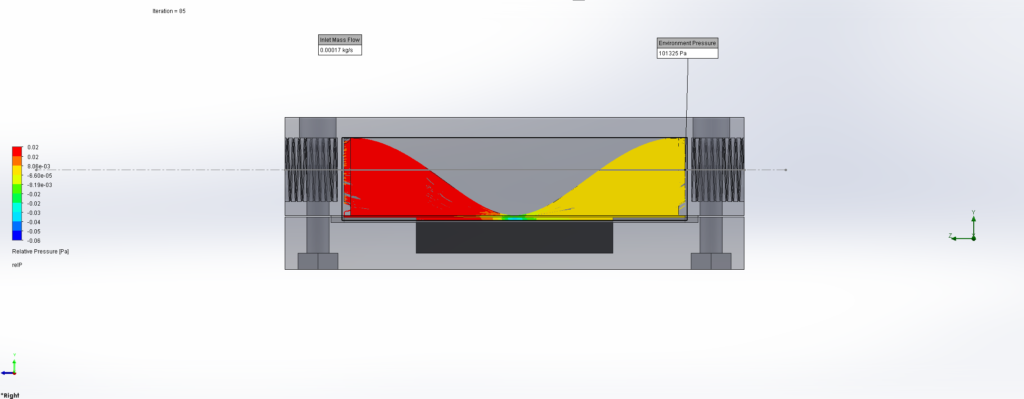
Right 
Isometric
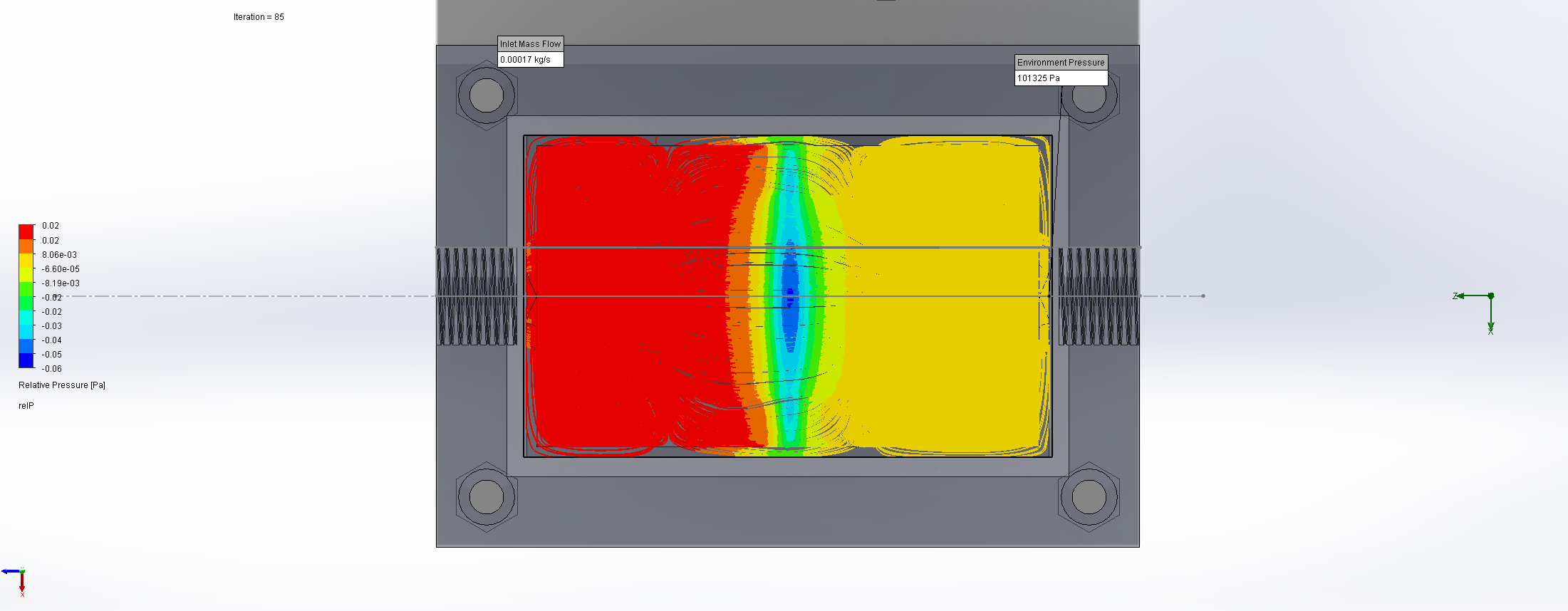
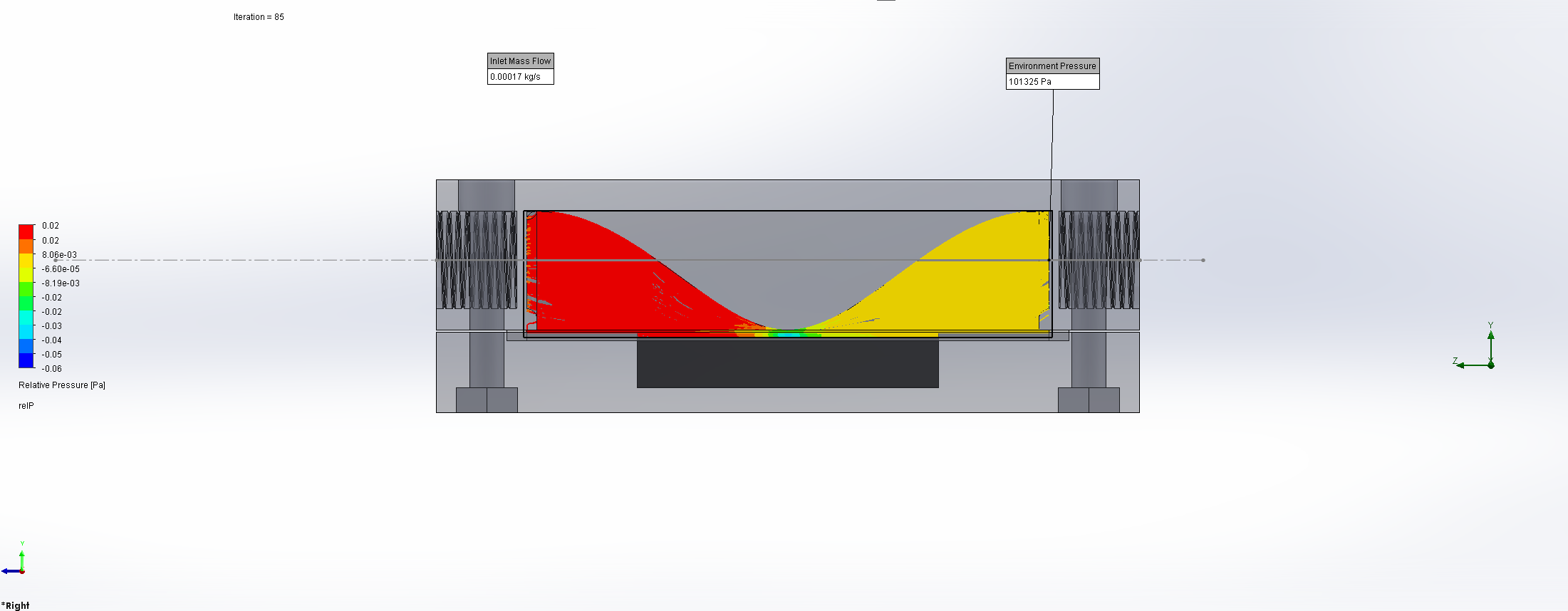

The simulation shows that we successful designed a chamber with a defined low-pressure area through utilization of the Venturi effect. Furthermore, the chamber features a mostly unobstructed flow path. Vortex formation can be observed, but the vortices are non-overlapping and localized to the four corners of the chamber, resulting in a clear path through the middle of the chamber, right through the measurement zone. This establishes a degree of certainty that the majority of separated bacteria leave the chamber with the flow and are accounted for in the measurement process.
After we were reasonably certain that our FlowChamber had the potential to produce good and repeatable results, we printed the chamber.
The CAD files, assembly instructions and the used assay protocol can be found here.
Theory – SnapGene
For a proof of concept of a stable co-culture we needed a method to proof that those differently modified cells indeed can live together – like roommates – and form a relatively homogenous biofilm. The method of our choice to test whether we were able to create a co-culture was fluorescence microscopy. By introducing different fluorescence proteins as strong markers, the successful cultivation of a co-culture can be validated.
To be able to perform our assay we had to design vectors that carry the fluorescence proteins.
We chose the plasmid pDG3661 as our backbone, because it features the necessary flanking regions for genomic integration into the amyE-Locus via double crossover homologous recombination36, as well as different antibiotic resistances which we could use to screen for a successful transformation. Furthermore, it can also be used in E. coli, which would enable us to amplify the vector before transforming it into B. subtilis.37
For the in silico planning of the vector, we followed these two papers: Veening et al. 200938 and van Gestel et al. 201435, which led to some difficulties in the form of not published sequences. To approximate the pJWV017 plasmid38 we designed two primers, pDG3661-Front and pDG3661-Back (Table 1), which linearized the pDG3661 plasmid, removed the existing spoVG-lacZ gene and also introduced a FseI restriction site.
Circularization of the previously created linear fragment results in our pJWV017 analogon (Figure 10), which features the restriction sites necessary for further handling.
After the creation of the pJWVO17 analogon, we deviated from the instructions of the Veening paper38 and began following the van Gestel paper35, because we wanted a promoter that constitutively expressed our insert in B. subtilis, something the rrnB-promoter described in the Veening paper38 was not able to accomplish, but the in van Gestel’s paper35 mentioned hyperspank-promoter could.
Encountering yet another not published Sequence (pDG111), we improvised: we used the Primers oHYSPANK1 and oHYSPANK2 described by van Gestel35 to clone the hyperspank promoter from the phyGFP plasmid, whose plasmid map we kindly got gifted from Prof. Dr. Ákos T. Kovács. This resulted in the same promoter that van Gestel et al. 2014 had received from the restriction and amplification out of the pDG111 plasmid.35
Using an overhang PCR to add a NheI and FseI restriction sites to the front and back of each of our two-fluorescence markers mKATE2 and Cycle-3-GFP, we then were able to assemble our final vectors using restriction ligation cloning, restricted by the enzymes EcoRI, NheI and FseI, resulting in our final Vector (Figure 11).


Table 1. List of used primers
| pDG3661-Front | 5’-GGCCGGCCAATAACCGGATCCTAGGACGCC-3’ |
| pDG3661-Back | 5’-GAATTCTCATGTTTGACAGCTTATCATCGG-3’ |
| oHYSPANK135 | 5’-CACGGAATTCAAACGAAAGGCTCAGTC-3′ |
| oHYSPANK235 | 5’-CGACGCTAGCCATTTCCTCTCCTCCTTAAGC-3′ |
Lab
Analyzing methods
Colony architecture analysis
To be able to conduct the colony architecture analysis of the biofilms, it was necessary to grow them at the air-agar interface. Therefore, 10-20 µl of the liquid overnight cultures, incubated in LB 5 medium, of:
- both of the GFP and mKATE labelled variants of the strain DK1042;
- the unlabelled variant of the strain DK1042, a NCIB3610 ComI Q12L mutant, which will from hereafter only be referred to as the ComI variant;
- the cultivated co-culture of both fluorescence labelled versions of DK1042;
- the negative control in form of LB 5 media
were plated out with glass beads onto minimal MSgg medium fortified with 1.5% Bacto agar and afterwards incubated for 3 days at 25°C.1
After the incubation period the formed B. subtilis biofilm could either be used for the examination of its stability and the influence of different environmental conditions (pH level, temperature, etc.) with the FlowChamber or for the analysis of its colony architecture.
For the analysis of the colony architecture, the surface texture of the formed biofilm had to be examined. If the surface seemed to be rough, wrinkly or crinkly it is a clear indication that a good quality biofilm had emerged.39
See our lab results for further information.
Fluorescence analysis
To conduct the fluorescence analysis of the biofilms according to Kovács et al. 201435, it was necessary to grow them at the air-liquid interface in a 96-well plate.40 For the cultivation of the pellicle biofilms 10µl of the liquid overnight co-culture cultures were added to each well of the 96-well plate.40 Afterwards 90 µl minimal MSgg medium were added to each well containing a sample of the overnight culture.40 Then the 96-well plate was incubated for 48h to 72h at room temperature or 37°C under static conditions. The initial protocol we followed stated a temperature of 30°C.40 Since we were not equipped with an incubator with a temperature 30°C we used this variation.
For the fluorescence analysis of the emerged biofilm, the 96-well plate was examined under a fluorescence microscope.

For the recorded images a filter mask with 50px radius was used to determine the randomness of the arrangement.35

Adjacently the degree of assortment within one of the squares of the resulting grid will be determined:
- if the green and red fluorescence cells within the square(s) are spatially separated the degree of assortment is 1,
- if the green and red fluorescence cells are randomly mixed and have no distinct pattern the degree of assortment is 0,
- and if the green and red fluorescence cells form an alternating pattern the degree of assortment is –1. 35
See our lab results for further information.
Microscopy
With the BZ-X800 all-in-one fluorescence microscope
Liquid cultures
For the liquid mono-cultures 3 ml of LB 5 media were inoculated with 10 µl of the GFP labelled variant of DK1042, the mKATE labelled variant of DK1042 and the ComI variant from overnight liquid cultures. Afterwards the inoculated liquid cell cultures were incubated over night at 37°C in a shaker (150 rpm).40
For the liquid co-culture 6 ml of liquid LB 5 media were inoculated with 3 µl of the GFP labelled and 3 µl of the mKATE variants of DK1042 from overnight liquid cultures of both fluorescence labeled DK1042 variants.41 Afterwards the inoculated liquid cell cultures were incubated overnight at 37°C in a shaker (150 rpm).41
The next day, 600 to 800 µl of the fresh liquid overnight cultures were transferred to a microscope slide and analyzed afterwards.
For the liquid culture analysis by microscopy, a Keyence BZ-X800 all-in-one fluorescence microscope with a high-resolution 2.8-megapixel monochrome charge-coupled device (CCD) sensor was used.42 The CCD allows an accurate one-click transition between monochrome and high-resolution color observation by using a color filter.42
The red fluorescence protein mKATE was visualized with a BZ-X TRITC filter cube (model OP-87764; excitation wavelength 525 to 545 nm; emission wavelength 605 to 670 nm; wavelength of the dichroitic mirror 565 nm).43 Green fluorescent proteins (GFP) were visualized with a BZ-X GFP filter cube (model OP-87763; excitation wavelength 440 to 470 nm; emission wavelength 525 to 550nm; wavelength of the dichroitic mirror 495 nm).44
Microscopy pictures were taken with the PlanApo_λ 20x 0.75 and the PlanApo_λ 60x 1.40 lenses in the multi- and single-color mode using BZ-X800 Viewer software.42,45 First, the reference photos of the ComI variant were taken in the multi color mode: a picture of the cells in the brightfield, pictures of the cells under both fluorescence filters separately and a picture of the overlay of both fluorescence filters (Figure 16 and Figure 17). Under the same conditions pictures of GFP and mKATE labeled variants of DK4210 were taken in the brightfield as well as under the fluorescence filters in the single-color mode (Figure 16 and Figure 17). Lastly, pictures of the cells of the co-cultures containing the GFP and mKATE labeled variants of DK1042 were taken in the multi-colored mode (Figure 18). These included an image of the cells in brightfield, pictures of the cells under both fluorescence filters and a picture of the overlay. If there was any form of motion visible in the liquid cultures additionally a video of the live picture shown in the BZ-X800 Viewer software42,45 was taken.
See our lab results for further information.
Biofilms

For the cultivation of the biofilms 10µl of the overnight liquid cultures were added in to each well of a 96-well plate40 according to the distribution scheme shown above. Afterwards 90µl minimal MSgg medium was added to each well containing a sample of the overnight culture.40 Now the 96-well plate was incubated for 48h to 72h at 30°C or at 37°C under static conditions.40
The biofilms that were grown in the 96-well plates were transferred to a microscope slide and analyzed.
For the biofilm analysis by microscopy, a Keyence BZ-X800 all-in-one fluorescence microscope with a high-resolution 2.8-megapixel monochrome charge-coupled device (CCD) sensor was used.42 The CCD allows an accurate one-click transition between monochrome and high-resolution color observation by using a color filter.42
The red fluorescence protein mKATE was visualized with a BZ-X TRITC filter cube (model OP-87764; excitation wavelength 525 to 545 nm; emission wavelength 605 to 670nm; wavelength of the dichroitic mirror 565nm).43 Green fluorescent protein (GFP) was visualized with a BZ-X GFP filter cube (model OP-87763; excitation wavelength 440 to 470 nm; emission wavelength 525 to 550nm; wavelength of the dichroitic mirror 495nm).44
Microscope pictures were taken and analyzed as described in the microscopic examination of the liquid cultures (Figures 19-26).
See our lab results for further information.
With the Operetta High Content Imaging System

The cultivation of the biofilms was executed as described in the microscopic examination of the biofilms with the BZ-X800 all-in-one fluorescence microscope.
For the analysis of the grown biofilm, we were able to use the Operetta High Content Imaging System from PerkinElmer 46,47 at the Cardoso lab at the Technical University of Darmstadt under the supervision of Dr. Alexander Rapp (Figure 27).
See our lab results for further information.
FlowChamber
We not only designed and printed our FlowChamber, but also wrote a protocol that ensures repeatable results. Interested? Check it out on our Contributions page!
Results
While our time in the lab was very limited, we were extremely lucky to receive variants of the B. subtilis strain DK1042 already labelled with the fluorescence proteins GFP and mKATE. These are originating from the plasmids phyGFP and phymKATE2. Also, we received an unlabeled variant of DK1042, which is a NCIB3610 ComIQ12L mutant. From here on, it will only be referred to as “ComI variant”.
These were kindly gifted by Prof. Dr. Ákos Kovács from the Technical University of Denmark. We chose to work directly with those variants of DK1042. With those we simulated and tested our multi-functional co-culture and biofilm in the lab.
For the analysis of the cultivated co-culture and the formed multi-functional pellicle biofilm different aspects like homogeneity, stability and quality were tested in various experiments.
Liquid cultures:
Firstly, we examined whether the received variants of DK1042 showed the expected fluorescence. For this, we analyzed the inoculated liquid cultures – mono and co-culture – with a fluorescence microscope (Figures 16-18).
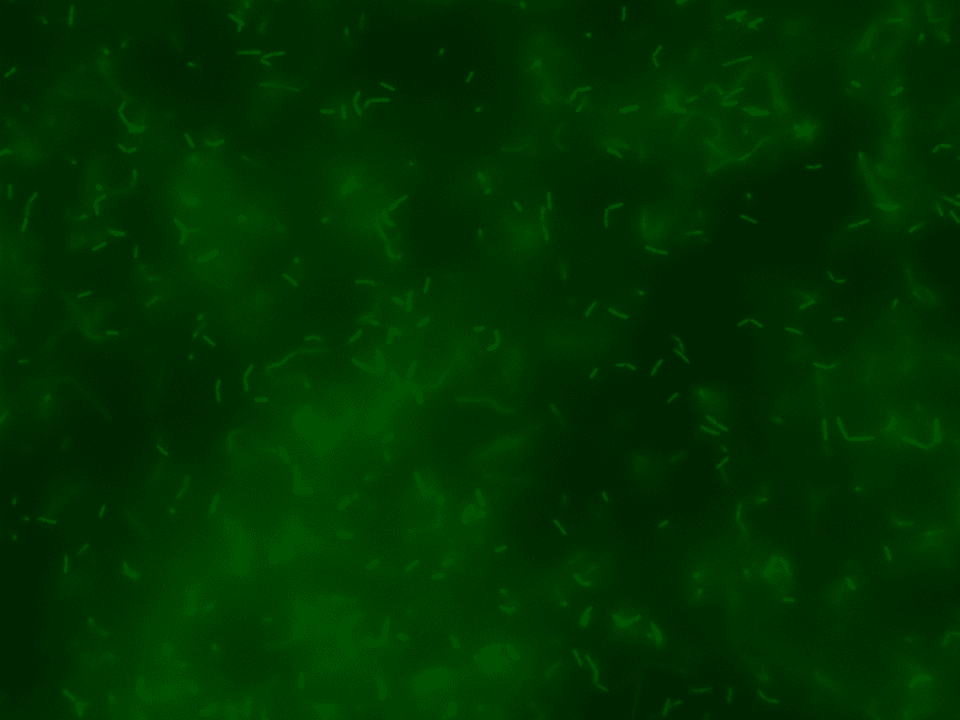
GFP in GFP spectrum at 60x 
NCIB3610 – ComI Brightfield at 60x 
NCIB3610 – ComI in GFP spectrum at 60x

mKate in TxRed spectrum at 60x NCIB3610 – ComI Brightfield at 60x 
NCIB3610 – ComI in TxRed spectrum at 60x
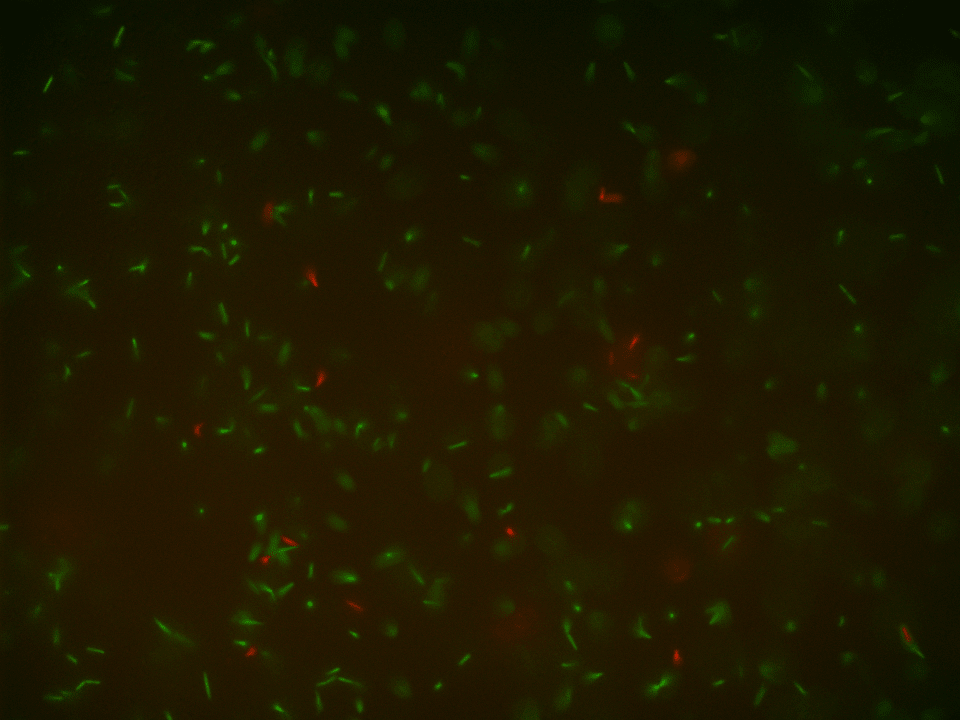
GFP + mKATE CoCulture at 60x – in GFP and in TxRed spectrum 
NCIB3610 – ComI Brightfield at 60x 
NCIB3610 – ComI in GFP spectrum at 60x 
NCIB3610 – ComI in TxRed spectrum at 60x
The pictures of the liquid mono-cultures were taken with the BZ-X800 all in one fluorescence microscope from Keyence using the PlanApo_λ 60x 1.40 lens in the single-color mode.42,45 The pictures of the liquid co-cultures were also taken with the BZ-X800 all in one fluorescence microscope from Keyence using the PlanApo_λ 60x 1.40 lens but in the multi-color mode.42,45 They clearly display that the production of the fluorescence proteins worked correctly in both variants in their mono-cultures and in the co-culture.
As a reference, a picture of the unlabeled variant of DK1042, which is a NCIB3610 ComI Q12L mutant, was taken under the exact same conditions meaning same lens and same settings. As expected, the ComI variant did not show any kind of fluorescence.
Biofilm analysis under the fluorescence microscope BZ-X90042,45 :
After proving that the received variants of DK1042 showed the expected fluorescence, we went on growing biofilms. We grew biofilms of all three of our mono-cultures and one co-culture for our analyses. For the cultivation of our pellicle biofilms in 96-well plates we used the biofilm-promoting nutritive medium Msgg (minimal salts glycerol glutamate)40 and the instructions on biofilm cultivation we had received from Prof. Dr. Ákos Kovács as well.
In the sections of all three mono-cultures (GFP, mKATE and ComI) as well as the co-culture of GFP and mKATE a pellicle biofilm was formed at the air-liquid interface.
No visible differences in texture, color or amount of biofilm could be observed from the comparison of the four well-columns with emerged biofilms.
For more in depth analyses the biofilms were transferred carefully from their single wells of the 96-well plate onto a microscope slide and analyzed under the BZ-X800 all in one fluorescence microscope from Keyence (Figures 19-26).

GFP biofilm in GFP spectrum at 60x 
NCIB3610 – ComI biofilm Brightfield at 60x 
NCIB3610 – ComI in GFP spectrum at 60x 
NCIB3610 – ComI in TxRed spectrum at 60x
mKATE biofilm in TxRed spectrum at 60x NCIB3610 – ComI biofilm Brightfield 60x NCIB3610 – ComI in TxRed spectrum at 60x 60x NCIB3610 – ComI in GFP spectrum at 60x
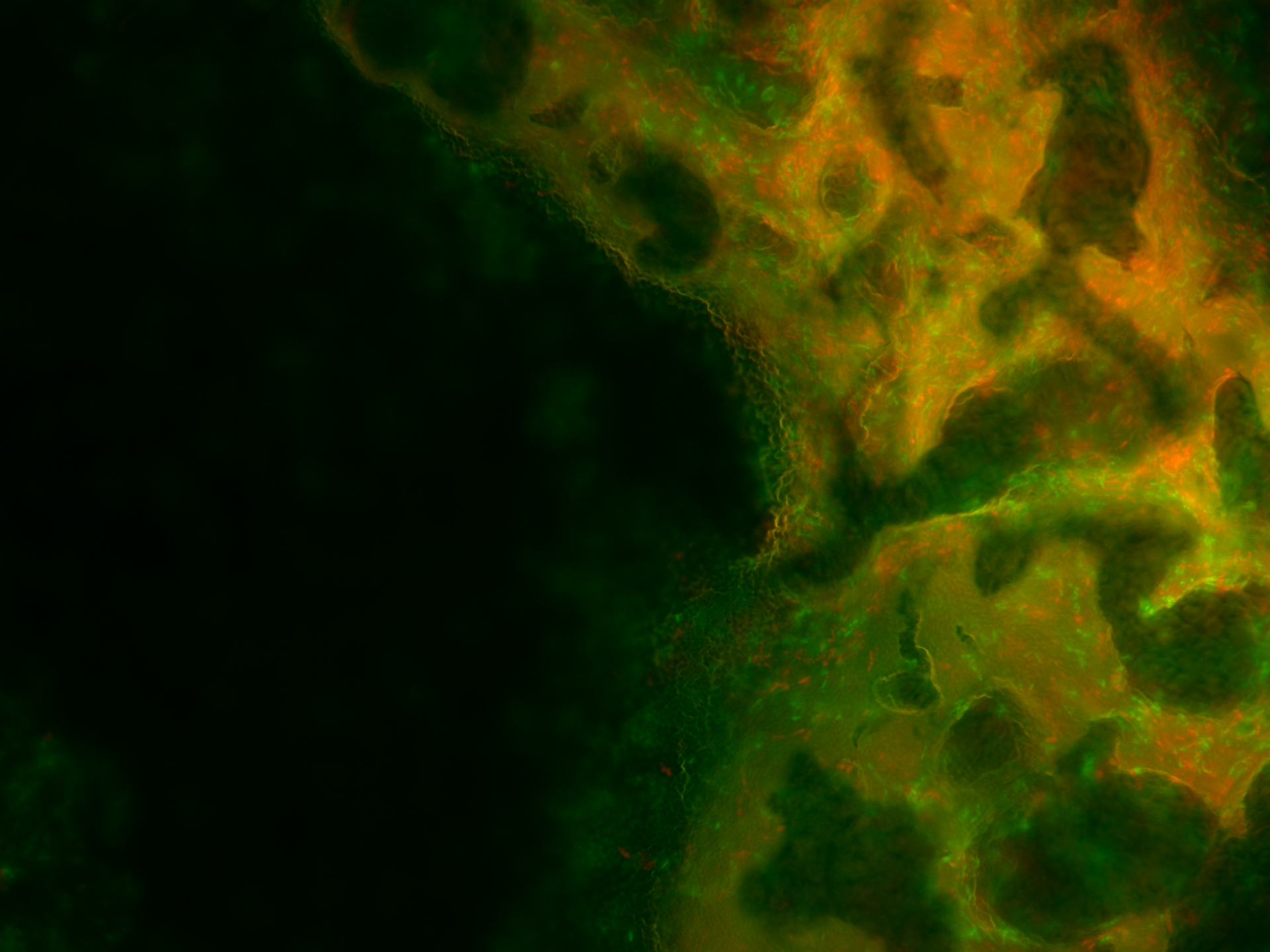
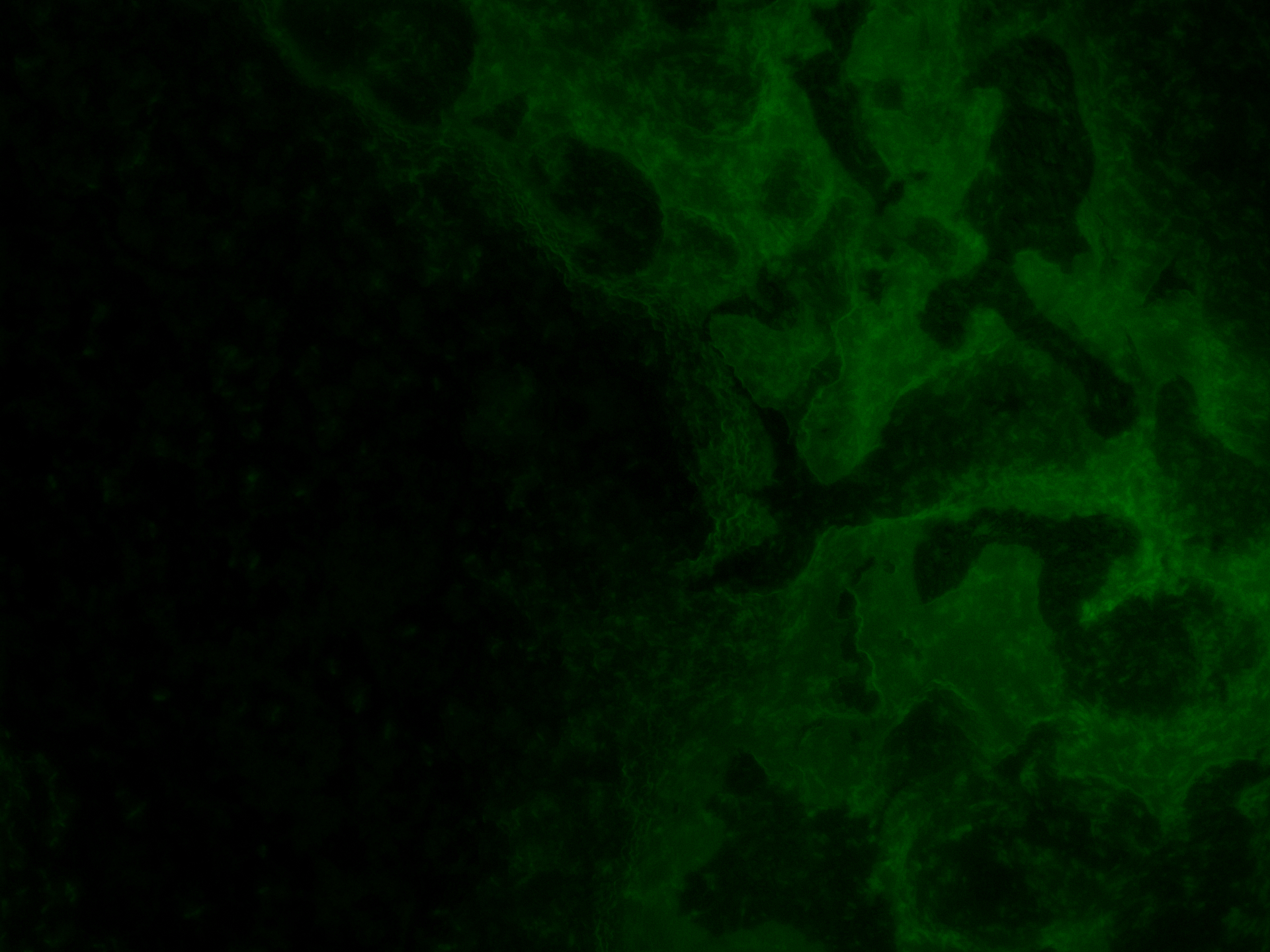
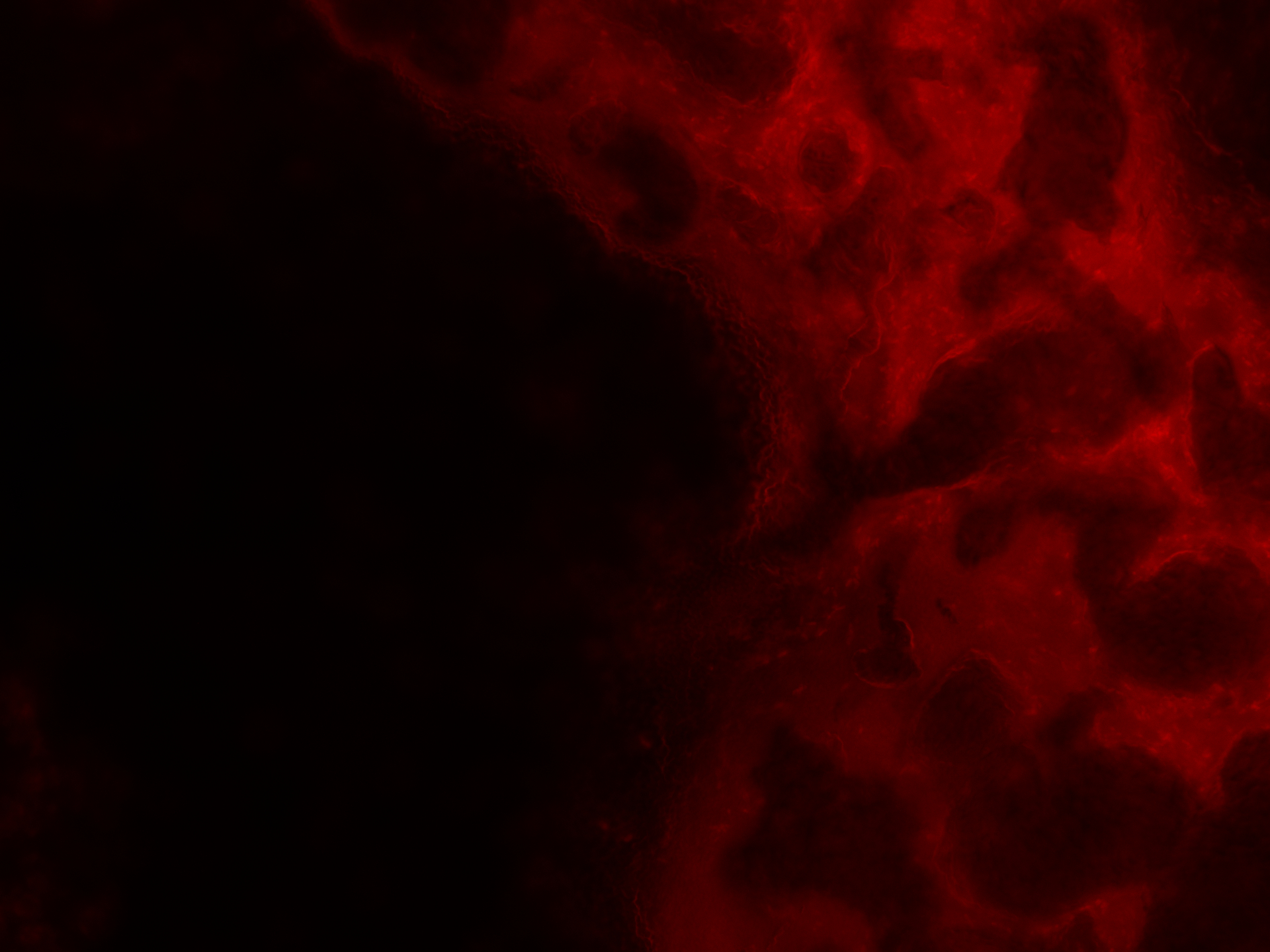


As for the analysis the biofilm was moved to the microscope slides by hand, even though we tried to leave it as untouched as possible, the homogeneity could have been impaired during this process. Therefore, a distribution analysis of the multi-functional biofilm as described in Kovács et al. 201435 was not possible. There, it was used to determine the degree of assortment of the cells in the biofilm which in turn was used for evaluation of its degree of homogeneity.
However, with the bare eye the cells in the multi-functional biofilm seemed to be evenly distributed on the microscopy images. In our opinion, it is very unlikely that this degree of homogeneity could have been achieved by the manual transfer of the biofilm.
Biofilm analysis under the Operetta High Content Imaging System46,47:
We were not satisfied with the pictures that we were able to take with the BZ-X800 microscope42,45. Especially because we were not able to conduct the distribution analysis of the multi-functional biofilm to determine the degree of assortment of the cells in the biofilm, to evaluate its degree of homogeneity (Kovács et al 201435 ).
Therefore, we decided to try a distribution analysis again, but this time with a microscope that didn’t require us to manually transfer the emerged biofilm samples from the 96-well plate to a microscope slide. We were able to use the Operetta High Content Imaging System from PerkinElmer46,47 at the Cardoso lab at the Technical University of Darmstadt under supervision of Dr. Alexander Rapp. The advantage of this microscope is that the 96-well plate containing the different biofilm samples could be simply placed in the microscope and analyzed afterwards.
As in our first attempt for distribution analyses we grew biofilms of four samples: the mono-cultures (GFP, mKATE and ComI variant) and the co-culture. Our pellicle biofilms were cultivated in a 96-well plate containing a special glass base, which was kindly provided to us by Dr. Alexander Rapp. We used the biofilm-promoting nutritive medium Msgg (minimal salts glycerol glutamate)40 and the instructions on biofilm cultivation we had received from Prof. Dr. Ákos Kovács.
In the wells of all three mono-cultures (GFP, mKATE and ComI variant) as well as in the wells of the co-culture pellicle biofilms were formed at the air-liquid interface. Again, no visible differences in texture, color or amount of biofilm could be observed from the comparison of the four well columns with emerged biofilms by the bare eye.
For an in depth analyses the biofilms in the 96-well plate were examined with the Operetta High Content Imaging System46,47 with the help of Dr. Alexander Rapp.
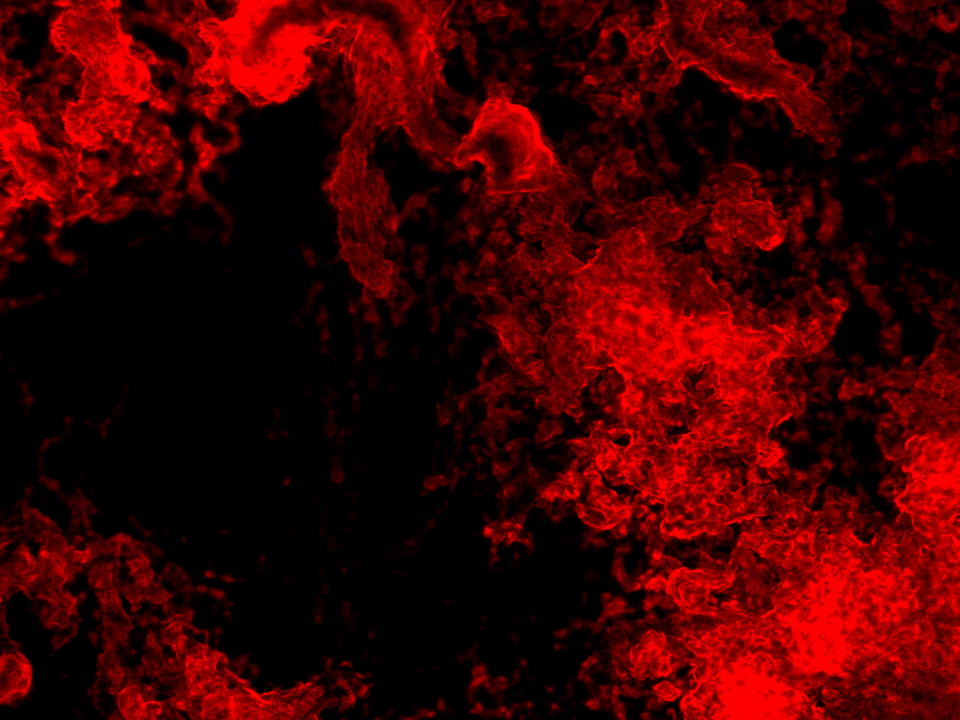
Even though this way the microscopy images were significantly better, an exact distribution analysis of the multi-functional biofilms was not possible because no single cells or cell colonies could be visualized. As can been seen in Figure 27 the extracellular matrix formed around the cells in the multi-functional biofilm as well as around the cells in the mono-cultures were too thick and not transparent making it impossible to take a look at the cell distribution beneath it.
Still, with the naked eye a distinct color pattern was noticeable in the shot of the extracellular matrix of the multi-functional biofilm (see row 6 in the Figure 27 highlighted with a pink box).

It can be clearly seen that both fluorescence labelled variants of DK1042 were part of the emerged biofilm in the column 6 of Figure 27. At this level the distribution seemed to be fairly even. Whether this would also have been the case at a cellular level is pure speculation. This confirmed what we and our experts already believed. The familiarity of the genetically modified B. subtilis cells indeed is sufficient to ensure the formation of a stable, homogenous co-culture – at least at higher level of cell distribution observation.
Biofilm Colony Architecture Analysis:
After three-day incubation of DK1042 with GFP or mKATE, the ComI variant as well as the co-culture on Msgg agar plates a colony architecture analysis was performed. As negative control pure LB 5 medium was used.
The biofilms that emerged at the air-liquid interface in the Petri dishes can be seen in Figure 28 that shows pictures of the biofilm from a distance and in Figure 29 that presents close-up photographs.
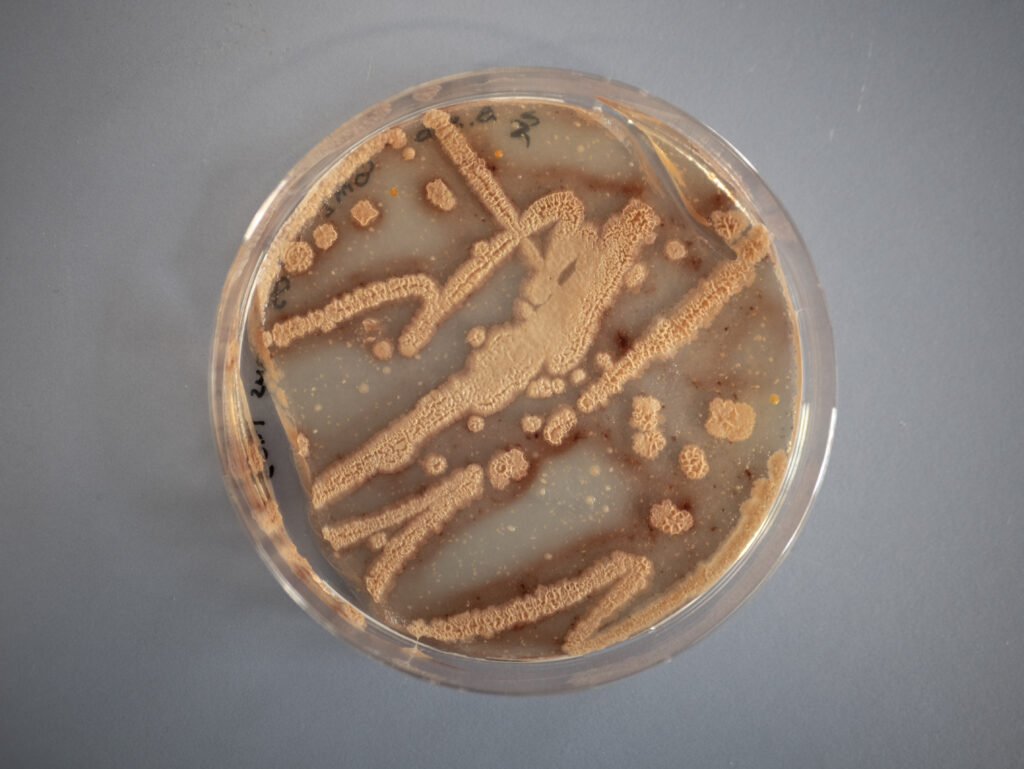
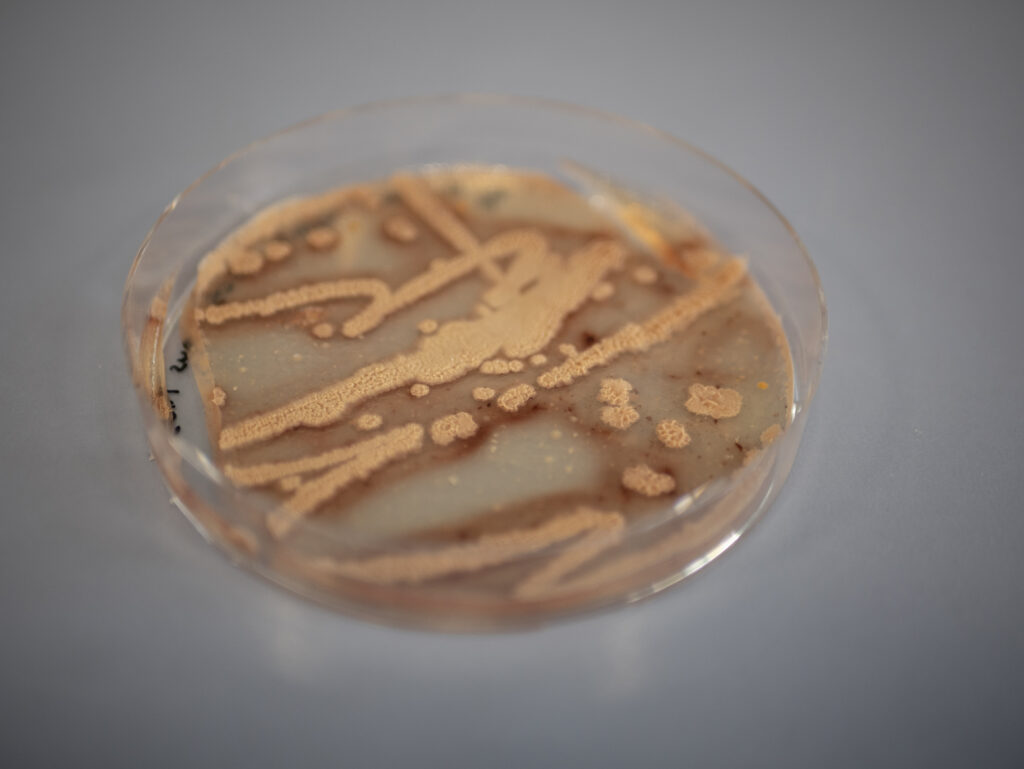
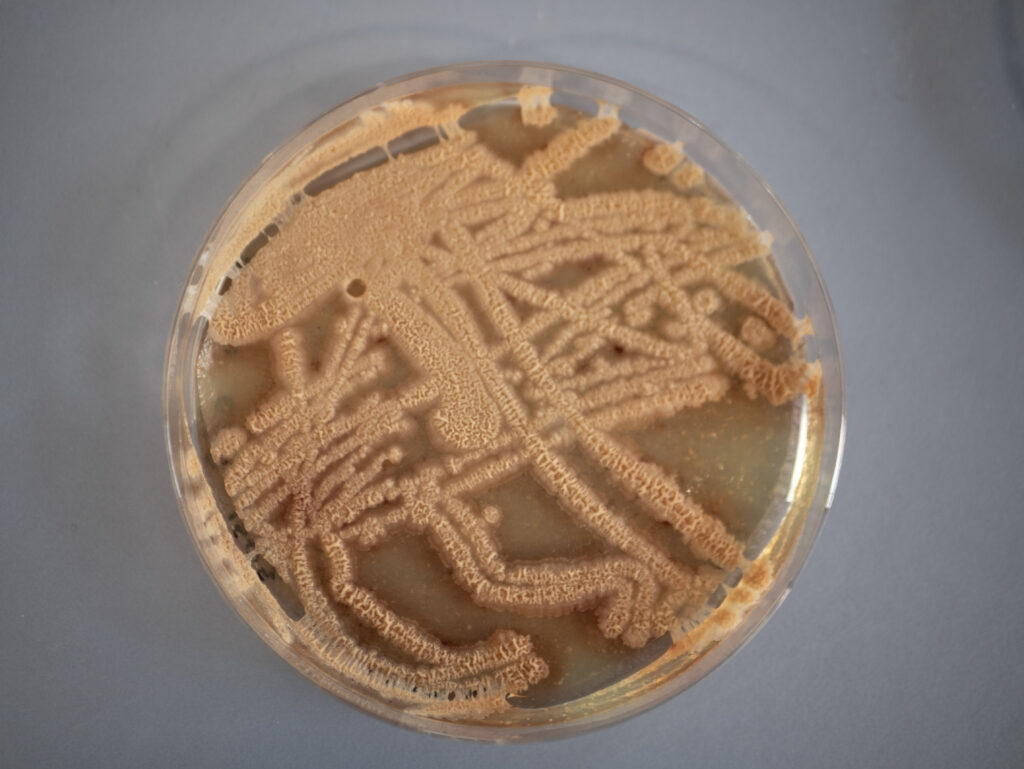



The aim of this analysis was examining the surface texture. In the pictures shown in the Figures 28 and 29, it can be seen that the surface seems rough, wrinkly or crinkly which is an indicator that an excellent biofilm emerged.39
No visibly noticeable differences in texture, color or amount of biofilm could be observed from the comparison of the four emerged biofilms with the bare eye. All showed the typical rough, wrinkly or crinkly surface indicating that an excellent biofilm emerged from the ComI variant, the GFP and mKATE variants of DK1042 as well as from the co-culture. As expected, there was no biofilm growth or any other kind of growth on the plate of the negative control.
Biofilms analysis with FlowChamber:
We designed a FlowChamber to analyze the stability of a biofilm. It allows us to expose the biofilm to different flow conditions and simulating different environments. If you are interested to find out how exactly we designed it and how we performed our assays take a look at our Contributions page.
Since our time in the laboratory was limited to 8 weeks from the middle of August until the middle of October due to COVID-19 regulations, we were not able to test our multi-functional biofilm’s stability with the FlowChamber. Besides the long delivery time of the chamber, we had to prioritize other aspects of our project such as the sensing circuit.
Conclusion:
These results indicate that our cultivated co-culture and the biofilm that emerged from it were stable and different cells within were evenly distributed – at least at a higher level of cell observation. This is as expected from the findings of our initial research phase as well as from conversations with our three experts Prof. Daniel Kearns, Dr. Ilka Bischofs and Prof. Dr. Ákos Kovács. For an analysis of the cell distribution at a cellular level an even better microscope, a different analyzing method and of course more time in the lab would have been necessary. Still, there seems to be no apparent need to resort to one or more of the three methods studied to synthetically produce a stable co-culture.
This successful creation of a co-culture biofilm signifies also the successful creation of the scaffold for the implementation of our modular defense-system. A potential user of PHIRE BYRD can just acquire his or her desired B. subtilis sleeper cells that responds to the presence of a specific pathogen with the expression of phages. These can then be cultivated alongside the functionalized cells they are supposed to protect forming a co-culture. When exposed to the proper conditions, given to you by us in a detailed protocol, a multi-functional biofilm will emerge that enables the existence of a safe, functionalized biofilm.
Outlook
The overall aim of this element of the project PHIRE BYRD in the lab was to create a multi‑functional Bacillus subtilis biofilm that contains and protects our sleeper cells and analyze its characteristics like the homogeneity, the stability and its quality. Thereby, we wanted to determine whether a multi-functional biofilm containing B. subtilis cells with different phage genomes could be achieved and would be stable and homogeneously distributed. To examine the homogeneous cell distribution in the biofilm, we planned to perform a fluorescence assay determining the overall distribution of the fluorescence labelled cells and carry out quantifications via a distribution analysis. The quality of the emerged biofilm was supposed to be appraised via macroscopically visible parameters like the texture of the biofilm and the overall appearance. To assess the stability of the multi-functional biofilm a FlowChamber should have been used.
Altogether we were able to analyze multiple biofilm samples with the desired analyzing methods:
- we tested whether the received variants of the strain DK1042 showed the expected fluorescence.
- we analyzed the biofilm samples with the BZ-X800 all in one fluorescence microscope from Keyence.42,45
- we analyzed the biofilm samples with the Operetta High Content Imaging System from PerkinElmer46,47 at the Cardoso lab at the Technical University of Darmstadt under supervision of Dr. Alexander Rapp.
- we performed a colony architecture analysis.
Since our time in the laboratory was limited to 8 weeks from the middle of August until the middle of October due to COVID-19 regulations, we were not able to analyze the biofilm samples to the extent we would have wanted to. This became evident in the following ways:
- we were not able to conduct the distribution analysis following the instructions from Kovács et al. 2014, due to the quality of the pictures we were able to take with the fluorescence microscope.
- we were not able to test our multi-functional biofilm’s stability with the FlowChamber. Besides the long delivery time of the chamber, we had to prioritize other aspects of our project such as the sensing circuit.
Still, we were able to confirm some of our expectations from our initial research and expert talks with Prof. Daniel Kearns, Dr. Ilka Bischofs and Prof. Dr. Ákos Kovács. The analyses that we were able to conduct indicated that our cultivated co-culture and the biofilm emerging from it, were indeed stable. Additionally, the different cells within it were evenly distributed – at least at a superficial level.
For more detailed information about our analysis and the results, take a look at our analyzing methods and results section.
Further procedures in the lab would be:
- to take the necessary pictures for the distribution analysis to be able to qualitatively analyze the distribution of the cells. This would be necessary to determine whether our hypothesis, that no synthetic interdependency is necessary to ensure a stable and homogeneous co-culture and multi-functional biofilm, was correct or not.
- to conduct stability analyses with our FlowChamber.
- to test whether a co-culture of cells from at least two differently modified B. subtilis with bacteriophage genomes can be formed.
- to test the formation, the stability and the homogeneity of this co-culture and its biofilm that contains cells from at least two differently modified B. subtilis with bacteriophage genomes.
References
- 1. Konkol MA, Blair KM, Kearns DB. Plasmid-Encoded ComI Inhibits Competence in the Ancestral 3610 Strain of Bacillus subtilis. Journal of Bacteriology. 2013:4085–4093. http://dx.doi.org/10.1128/JB.00696-13. doi:10.1128/jb.00696-13
- 2. Liu L, Liu Y, Shin H, Chen RR, Wang NS, Li J, Du G, Chen J. Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology. Applied Microbiology and Biotechnology. 2013:6113–6127. http://dx.doi.org/10.1007/s00253-013-4960-4. doi:10.1007/s00253-013-4960-4
- 3. Clements LD, Miller BS, Streips UN. Comparative Growth Analysis of the Facultative Anaerobes Bacillus subtilis, Bacillus licheniformis, and Escherichia coli. Systematic and Applied Microbiology. 2002:284–286. http://dx.doi.org/10.1078/0723-2020-00108. doi:10.1078/0723-2020-00108
- 4. Nakano MM, Zuber P. ANAEROBIC GROWTH OF A “STRICT AEROBE” (BACILLUS SUBTILIS). Annual Review of Microbiology. 1998:165–190. http://dx.doi.org/10.1146/annurev.micro.52.1.165. doi:10.1146/annurev.micro.52.1.165
- 5. Vlamakis H, Chai Y, Beauregard P, Losick R, Kolter R. Sticking together: building a biofilm the Bacillus subtilis way. Nature Reviews Microbiology. 2013:157–168. http://dx.doi.org/10.1038/nrmicro2960. doi:10.1038/nrmicro2960
- 6. Microorganisms & Microbial-Derived Ingredients Used in Food (Partial List) . U.S. Food & Drug Administration. 2018 Apr 1 [accessed 2021 Oct 3]. https://www.fda.gov/food/generally-recognized-safe-gras/microorganisms-microbial-derived-ingredients-used-food-partial-list
- 7. Chen I, Dubnau D. DNA uptake during bacterial transformation. Nature Reviews Microbiology. 2004:241–249. http://dx.doi.org/10.1038/nrmicro844. doi:10.1038/nrmicro844
- 8. Flemming H-C, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nature Reviews Microbiology. 2016:563–575. http://dx.doi.org/10.1038/nrmicro.2016.94. doi:10.1038/nrmicro.2016.94
- 9. Flemming H-C, Wingender J. The biofilm matrix. Nature Reviews Microbiology. 2010:623–633. http://dx.doi.org/10.1038/nrmicro2415. doi:10.1038/nrmicro2415
- 10. Dunne WM Jr. Bacterial Adhesion: Seen Any Good Biofilms Lately? Clinical Microbiology Reviews. 2002:155–166. http://dx.doi.org/10.1128/CMR.15.2.155-166.2002. doi:10.1128/cmr.15.2.155-166.2002
- 11. Donlan RM. Biofilms: Microbial Life on Surfaces. Emerging Infectious Diseases. 2002:881–890. http://dx.doi.org/10.3201/eid0809.020063. doi:10.3201/eid0809.020063
- 12. Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Peptide Signaling in the Staphylococci. Chemical Reviews. 2010:117–151. http://dx.doi.org/10.1021/cr100370n. doi:10.1021/cr100370n
- 13. Rosenthal AZ, Qi Y, Hormoz S, Park J, Li SH-J, Elowitz MB. Metabolic interactions between dynamic bacterial subpopulations. eLife. 2018. http://dx.doi.org/10.7554/eLife.33099. doi:10.7554/elife.33099
- 14. Billings N, Birjiniuk A, Samad TS, Doyle PS, Ribbeck K. Material properties of biofilms—a review of methods for understanding permeability and mechanics. Reports on Progress in Physics. 2015:036601. http://dx.doi.org/10.1088/0034-4885/78/3/036601. doi:10.1088/0034-4885/78/3/036601
- 15. Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends in Microbiology. 2005:27–33. http://dx.doi.org/10.1016/j.tim.2004.11.007. doi:10.1016/j.tim.2004.11.007
- 16. Root Patch. iGEM Team Groningen. [accessed 2021 Oct 5]. https://2020.igem.org/Team:Groningen
- 17. B-TOX. iGEM Team TU-Darmstadt. [accessed 2021 Oct 5]. https://2020.igem.org/Team:TU_Darmstadt
- 18. Stephens K, Pozo M, Tsao C-Y, Hauk P, Bentley WE. Bacterial co-culture with cell signaling translator and growth controller modules for autonomously regulated culture composition. Nature Communications. 2019. http://dx.doi.org/10.1038/s41467-019-12027-6. doi:10.1038/s41467-019-12027-6
- 19. Lombardía E, Rovetto AJ, Arabolaza AL, Grau RR. A LuxS-Dependent Cell-to-Cell Language Regulates Social Behavior and Development in Bacillus subtilis. Journal of Bacteriology. 2006:4442–4452. http://dx.doi.org/10.1128/JB.00165-06. doi:10.1128/jb.00165-06
- 20. Duanis-Assaf D, Steinberg D, Chai Y, Shemesh M. The LuxS Based Quorum Sensing Governs Lactose Induced Biofilm Formation by Bacillus subtilis. Frontiers in Microbiology. 2016. http://dx.doi.org/10.3389/fmicb.2015.01517. doi:10.3389/fmicb.2015.01517
- 21. Bareia T, Pollak S, Eldar A. Self-sensing in Bacillus subtilis quorum-sensing systems. Nature Microbiology. 2017:83–89. http://dx.doi.org/10.1038/s41564-017-0044-z. doi:10.1038/s41564-017-0044-z
- 22. Kalamara M, Spacapan M, Mandic-Mulec I, Stanley-Wall NR. Social behaviours byBacillus subtilis: quorum sensing, kin discrimination and beyond. Molecular Microbiology. 2018:863–878. http://dx.doi.org/10.1111/mmi.14127. doi:10.1111/mmi.14127
- 23. Hays SG, Patrick WG, Ziesack M, Oxman N, Silver PA. Better together: engineering and application of microbial symbioses. Current Opinion in Biotechnology. 2015:40–49. http://dx.doi.org/10.1016/j.copbio.2015.08.008. doi:10.1016/j.copbio.2015.08.008
- 24. Goers L, Freemont P, Polizzi KM. Co-culture systems and technologies: taking synthetic biology to the next level. Journal of The Royal Society Interface. 2014:20140065. http://dx.doi.org/10.1098/rsif.2014.0065. doi:10.1098/rsif.2014.0065
- 25. Zhou K, Qiao K, Edgar S, Stephanopoulos G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nature Biotechnology. 2015:377–383. http://dx.doi.org/10.1038/nbt.3095. doi:10.1038/nbt.3095
- 26. Liu J, Prindle A, Humphries J, Gabalda-Sagarra M, Asally M, Lee DD, Ly S, Garcia-Ojalvo J, Süel GM. Metabolic co-dependence gives rise to collective oscillations within biofilms. Nature. 2015:550–554. http://dx.doi.org/10.1038/nature14660. doi:10.1038/nature14660
- 27. Zander A. Generation of auxotrophic Bacillus subtilis strains. Jena: Friedrich-Schiller-Universität Jena; 2016. http://www.clib-jena.mpg.de/theses/ice/ICE16006.pdf
- 28. Sidiq KR, Chow MW, Zhao Z, Daniel RA. Alanine metabolism in Bacillus subtilis. molecular microbiology. 2020;115(4):739–757. https://onlinelibrary.wiley.com/doi/abs/10.1111/mmi.14640. doi:https://doi.org/10.1111/mmi.14640
- 29. Glass DS, Riedel-Kruse IH. A Synthetic Bacterial Cell-Cell Adhesion Toolbox for Programming Multicellular Morphologies and Patterns. Cell. 2018:649-658.e16. http://dx.doi.org/10.1016/j.cell.2018.06.041. doi:10.1016/j.cell.2018.06.041
- 30. Saier Lab Bioinformatics Group . 1.B.54 The Intimin/Invasin (Int/Inv) or Autotransporter-3 (AT-3) Family. Transporter Classification Database. [accessed 2021 Oct 5]. http://www.tcdb.org/search/result.php?tc=1.B.54
- 31. Burgess NK, Dao TP, Stanley AM, Fleming KG. β-Barrel Proteins That Reside in the Escherichia coli Outer Membrane in Vivo Demonstrate Varied Folding Behavior in Vitro. Journal of Biological Chemistry. 2008:26748–26758. http://dx.doi.org/10.1074/jbc.M802754200. doi:10.1074/jbc.m802754200
- 32. Chen C-L, Wu S-C, Tjia WM, Wang CLC, Lohka MJ, Wong S-L. Development of a LytE-based high-density surface display system inBacillus subtilis. Microbial Biotechnology. 2007:177–190. http://dx.doi.org/10.1111/j.1751-7915.2007.00017.x. doi:10.1111/j.1751-7915.2007.00017.x
- 33. Spirig T, Weiner EM, Clubb RT. Sortase enzymes in Gram-positive bacteria. Molecular Microbiology. 2011:1044–1059. http://dx.doi.org/10.1111/j.1365-2958.2011.07887.x. doi:10.1111/j.1365-2958.2011.07887.x
- 34. Muyldermans S. Nanobodies: Natural Single-Domain Antibodies. Annual Review of Biochemistry. 2013:775–797. http://dx.doi.org/10.1146/annurev-biochem-063011-092449. doi:10.1146/annurev-biochem-063011-092449
- 35. van Gestel J, Weissing FJ, Kuipers OP, Kovács ÁT. Density of founder cells affects spatial pattern formation and cooperation in Bacillus subtilis biofilms. The ISME Journal. 2014:2069–2079. http://dx.doi.org/10.1038/ismej.2014.52. doi:10.1038/ismej.2014.52
- 36. Kuzminov A. Homologous Recombination—Experimental Systems, Analysis, and Significance Lovett ST, Kuzminov A, editors. EcoSal Plus. 2011. http://dx.doi.org/10.1128/ecosalplus.7.2.6. doi:10.1128/ecosalplus.7.2.6
- 37. Krásný L, Gourse RL. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. The EMBO Journal. 2004:4473–4483. http://dx.doi.org/10.1038/sj.emboj.7600423. doi:10.1038/sj.emboj.7600423
- 38. Veening J-W, Murray H, Errington J. A mechanism for cell cycle regulation of sporulation initiation in Bacillus subtilis. Genes & Development. 2009:1959–1970. http://dx.doi.org/10.1101/gad.528209. doi:10.1101/gad.528209
- 39. Kearns D. Video conference with Daniel Kearns from the Indiana University Bloomington (IUB). Zoom conference room; 2021.
- 40. Kovács Á. Video Conference with Ákos Kovács from the Technical University Denmark. Zoom conference room; 2021.
- 41. Guo Y, Yang R, Zhang Z, Wang X, Ye X, Chen S. Synergy of carbon and nitrogen removal of a co-culture of two aerobic denitrifying bacterial strains, Acinetobacter sp. GA and Pseudomonas sp. GP. RSC Advances. 2018:21558–21565. http://dx.doi.org/10.1039/C8RA02721H. doi:10.1039/c8ra02721h
- 42. All-in-one Fluorescence MicroscopeBZ-X800/BZ-X810 User’s Manual. https://research.ncsu.edu. [accessed 2021 Oct 9]. https://research.ncsu.edu/cmif/files/2020/07/BZ-X800-Viewer-Users-Manual.pdf
- 43. BZ-X Filter TRITC. KEYENCE DEUTSCHLAND gmbH. [accessed 2021 Oct 9]. https://www.keyence.de/products/microscope/fluorescence-microscope/bz-x700/models/op-87764/
- 44. BZ-X Filter GFP. KEYENCE DEUTSCHLAND GmbH. [accessed 2021 Oct 9]. https://www.keyence.de/products/microscope/fluorescence-microscope/bz-x700/models/op-87763/
- 45. Kompaktes Fluoreszenzmikroskop BZ-X800. KEYENCE DEUTSCHLAND GmbH. [accessed 2021 Oct 18]. https://www.keyence.de/ss/products/microscope/bz-x800_long/
- 46. Operetta CLS High-Content Analysis System . PerkinElmer Inc. [accessed 2021 Oct 10]. https://www.perkinelmer.com/de/product/operetta-cls-system-hh16000000
- 47. Operetta_brochure. https://resources.perkinelmer.com. [accessed 2021 Oct 10]. https://resources.perkinelmer.com/lab-solutions/resources/docs/011034_01_BRO_Operetta_brochure.pdf