
Introduction
Our system PHIRE BYRD introduces an adaptive immune-like system to functionalized biofilms by triggering phage expression upon invasion of pathogens. Within our very limited lab time we managed to conduct proof of concept experiments showing the feasibility of the system after further DBTL optimization cycles.
Working on every part of our very comprehensive project in just eight weeks, we are proud to have successfully simulated a multi-functional biofilm co-culture and tested it for different aspects like homogeneity, stability and quality. We have assembled our pathogen sensing circuit using GoldenBraid 2.0 cloning. Functionality of our genetic switch controlling the induction of lambda phage expression was validated using a fluorescence reporter assay. By successfully cloning the cre gene into the pJUMP27_araC_pBAD backbone we did our first step to realize our kill-switch and thereby contributing to more biosafety.
Taken together, these results confirm the feasibility of our project, proving crucial parts of our responsive system with overlapping elements. These results highlight the mergeability of our certain parts to ultimately result in an applicable PHIRE BYRD system.
Biofilm
Creation of a Multi-functional Biofilm
The primary objective of this part of the project PHIRE BYRD is the creation of a biofilm which contains our sleeper cells. This biofilm is not only supposed to contain one kind of B. subtilis sleeper cell but multiple of different genetically modified B. subtilis sleeper cells from one strain DK1042.1 These sleeper cells each carry the genetic information of bacteriophages. In order to reduce the metabolic load on the B. subtilis cell and to maintain the modularity of our system, one sleeper cell can only carry the genome of one highly specialized bacteriophage that targets one specific pathogen. This way our emerging multi-functional B. subtilis biofilm will be a modular phage-defense system which ensures that it can be used against various pathogens. Additionally, the genetically modified cells in the grown biofilm should be distributed homogenously in order to achieve uniform protection. For the creation of a multi-functional biofilm, as a basis a stable multi-functional co-culture is needed.
In consultation with our experts on B. subtilis and biofilms, we agreed that the familiarity of the genetically modified B. subtilis cells should be sufficient to ensure the formation of a stable, homogenous co-culture and the introduction of synthetically engineered interdependencies therefore would not be necessary. Thus, it would be possible to mix different strains in the same ratio with another.
To simulate and test our multi-functional co-culture made up by multiple (at least two) differently genetically modified B. subtilis sleeper cells in the lab, we are using different fluorescence-labelled variants of the same strain DK1042. One contains the fluorescence protein GFP integrated in form of the plasmid phyGFP and the other the fluorescence protein mKATE integrated in form of the plasmid phymKate2.
For the analysis of the co-culture and the formed multi-functional pellicle biofilm different aspects like the homogeneity, the stability and their quality were tested:
- To examin the homogenous cell distribution in the biofilm, a fluorescence assay determining the overall distribution of the fluorescence labelled cells was conducted and quantified via a distribution analysis.
- The quality of the emerged biofilm was appraised through macroscopically visible parameters like the texture of the biofilms and its overall look.
- For the assessment of the stability of the multi-functional biofilm we used a FlowChamber.
In order to be able to conduct these analyses, samples of the overnight culture were distributed in biofilm-promoting nutritive medium Msgg in a 96-well plate in the following matter:

The green highlighted wells contain the GFP labelled variant of DK1042, the purple highlighted wells contain the mKATE labelled variant of DK1042, the pink highlighted wells the co-culture of both fluorescence labelled variants, the grey highlighted wells the unlabelled variant of the strain DK1042, a NCIB3610 ComI Q12L mutant, which will from hereafter only be referred to as the ComI variant and the light-grey highlighted wells the negative control in form of pure LB 5 media. The distribution is visualized in Figure 1.

Figure 2. Picture of the samples of the overnight culture in the 96-well plate, taken with the Operetta High Content Imaging System form Perkin Elmer at the Cardoso lab at TU Darmstadt. The well-column on the far left contains the GFP labelled variant of DK1042; the well-columns on the far right contains the mKATE labelled variant of DK1042; the well-column next to the column with the mKATE labelled variant of DK1042 contains the ComI variant; the well-column next to the column with the ComI variant contains the co-culture with both fluorescently labelled variants of DK1042 and the horizontal well-column on top of the well-plate contain the negative control in form of pure LB 5 media. In the microscope picture GFP, due to its higher fluorescent intensity, is that much enlightened because the contrast the pictures was set the same value to be able to compare them. Otherwise, pattern in co-culture would not have been visible.
As shown in the image taken of the 96-well-plate with the Operetta High Content Imaging System from PerkinElmer at the Cardoso lab at the Technical University of Darmstadt our co-culture and the biofilm emerging from it, are indeed stable and the different cells within it are evenly distributed as one can see with the bare eye due to a distinct color pattern being noticeable in the shot of the extracellular matrix of the multi-functional biofilm (see row 6 in the Figure 2highlighted with a pink box).
See our lab results for a more detailed evaluation of our proof of concept.
If any of these analyses had shown that the resulting co-culture and the biofilm emerging from it, is either not stable or the different cells within are not evenly distributed, one possibility would have been to resort to one or more of the three methodsstudied to synthetically produce a stable co-culture.
Pathogen Sensing
Pathogen Sensing via Allosteric Transcriptional Regulation
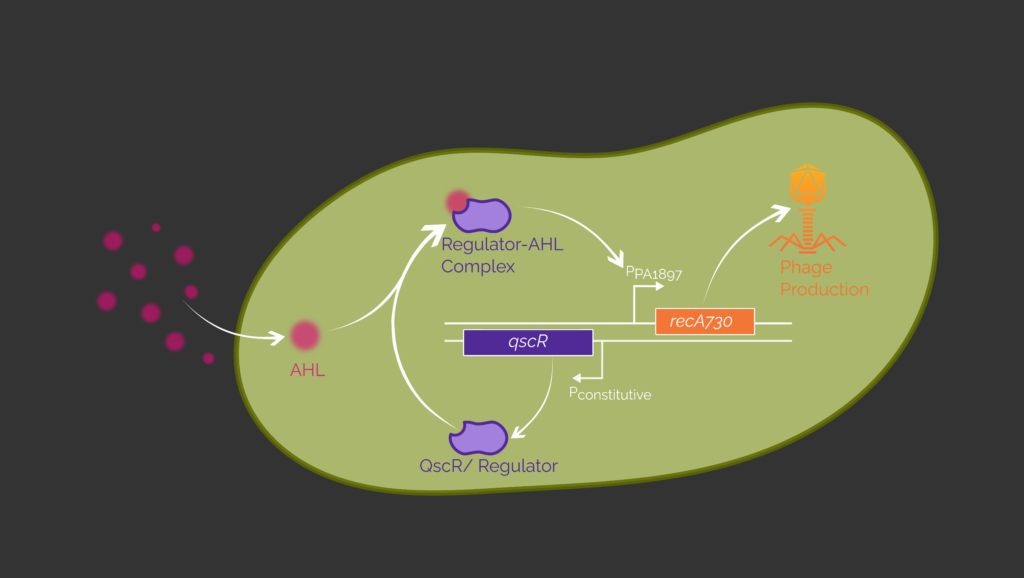
Our sensing circuit is designed to initiate phage production in the presence of the pathogen P. aeruginosa. The circuit is based on the allosteric transcription factor (aTF) QscR or LasR originating from the quorum sensing system of P. aeruginosa.2,3 Both aTF bind the acyl homoserine lactone (AHL) N-(3-oxododecanoyl)-homoserine lactone (3OC12-HSL) which is a quorum sensing autoinducer native to P. aeruginosa.4,5 The aTF are constitutively expressed and form a complex with 3OC12-HSL. This enables binding to its respective promoter. QscR binds to PPA1897 and LasR binds to PLux and therefore initiates transcription. With one of these inducible promoters we could later control the expression of recA730. (Figure 3) The protein RecA is responsible for the switch from the lysogenic to the lytic cycle of the lambda phage and thus for controlled phage production.6,7
As proof of concept, we use eGFP as a reporter protein instead of RecA730 to evaluate this sensing circuit by measuring fluorescence output in E. coli. (Figure 4) These E. coli cells are transformed with a plasmid containing the sensing circuit. Thereby, we reproduce already successful experiments by Wu et al. (2021) and Saide et al. (2011).2,3 We codon optimized the genes encoding for the aTF and eGFP for E. coli. The constitutive T7 promoter native to E. coli is used to control the expression of the aTF. In the lab we used the E. coli BL21 (DE3) strain, because these cells possess a T7 expression system. The T7 polymerase is expressed, if Isopropyl-β-D-thiogalactopyranosid (IPTG) is added to the culture medium.
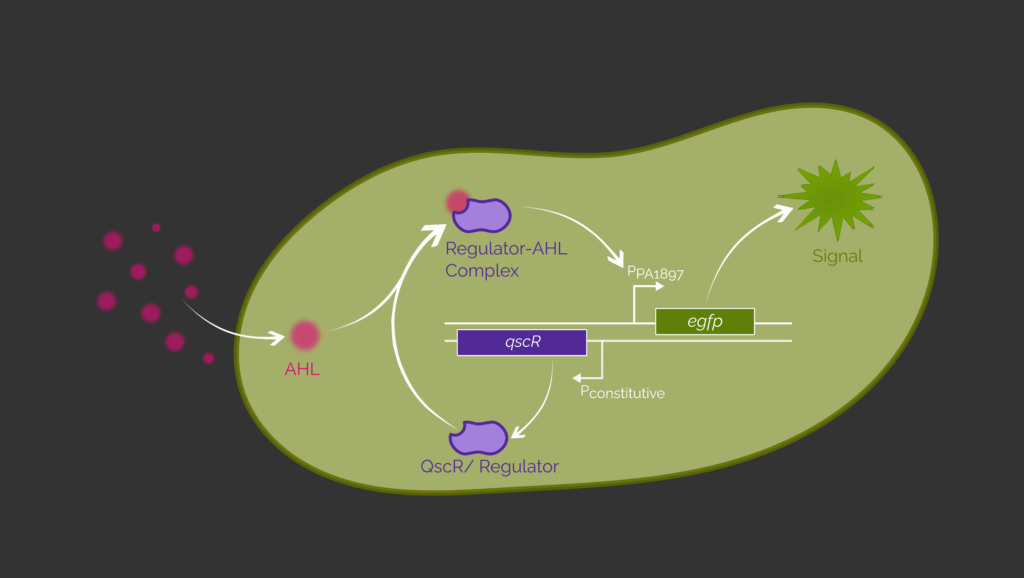
First, we evaluate the sensing circuit with QscR as the transcription factor. Therefore, transformed E. coli cells are incubated with no supplemented IPTG or 3OC12-HSL in the culture media. We expect no eGFP expression since the T7 polymerase and thus, QscR is not expressed.
The same result is expected when indeed IPTG but no 3OC12-HSL is added to the culture medium. Now, the T7 polymerase is present and the QscR gene is expressed. But since no AHL molecules entered the cell, the expression of eGFP cannot be activated.
In contrast, the presence of IPTG and 3OC12-HSL in the culture medium is expected to enable expression of eGFP and thus a detection of a fluorescence output. QscR is available in the cell due to transcription by the T7 polymerase. The AHLs in the culture enter the E. coli cell and form a complex with QscR. This enables the binding to its respective promoter PPA1897. Now, the expression of eGFP is possible and can be confirmed by fluorescence measurements.
The same experiments are performed with the sensing circuit containing the transcription factor LasR and its respective promoter PLux. If 3OC12-HSL molecules are present, the LasR-3OC12-HSL complex induces the promoter and egfp is expressed.
In this way, we are planned to prove that the expression of egfp is induced by adding 3OC12-HSL into the medium of transformed E. coli cells by using a simple fluorescence assay. This means the cells containing the sensing circuit respond to the presence of 3OC12-HSL by expression of egfp. Unfortunately, in the end we were not able to do all the experiments in the lab because the time was limited.
In order to respond with phage expression instead of fluorescence output, egfp must be replaced with recA730 which controls phage expression. This process is described in more detail in the next section.
Bacteriophages
Controlled Lytic Cycle Induction Utilizing a cI-RecA730 Based Genetic Switch
Our genetic switch activates phage induction when a quorum sensing molecule from an invading pathogen is detected. As proof of concept we want to show that our genetic switch is able to produce the fluorescent protein eGFP as a reporter for the induction of the lytic cylce in dependence of IPTG (representative for the quorum sensing molecule) as an inducer.
In our design bacteriophage genes as well as egfp are under control of the PR promoter which is repressed by cI, the lambda repressor. (Figure 5) The basic concept of our synthetic genetic switch is based on the two proteins RecA730 and TetR, which act as antagonists for the lambda repressor. The genes for both proteins are regulated by a T7 promoter.

For this experiment we use E. coli BL21(DE3) cells that contain an IPTG inducible T7 RNA polymerase. Prior to IPTG induction, recA730 and tetR will not be expressed and therefore cI is expressed constitutively. In presence of IPTG, the LacI repressor will detach from the lacUV5 promoter, enabling the expression of the T7 RNA polymerase. As a result, the TetR repressor binds to the PTetO promoter, blocking cI transcription. Furthermore, the additionally expressed recA730 cleaves the remaining cI, making the tripartite operator OR freely accessible, which results in egfp expression.
Since this promoter also maintains the native lysogenic state, we transformed E. coli BL21(DE3) and planned the transformation of the lysogenic strain E. coli MG1655(λ) with our synthetic construct as well. The fluorescent output is expected right around the time the lytic cycle in the lambda phage is induced which will result in cell death. Therefore, we measured the OD600 to monitor the cell number and the excitation at 485 nm to monitor the eGFP expression for the E. coli BL21(DE3) and due to the limited lab time, we did not get to perform the planned measurement of the lysogenic E. coli MG1655(λ) strain. IPTG induction should induce a fluorescent output in the BL21(DE3) strains while having minimal to no impact to the optical density. This is consistent with our results. (Figure 6) In comparison the lysogenic strain should show at around 45 min upon the induction a rapid decline in the optical density.6 This accounts for the time needed for the functional phage assembly and the cell lysis.

To further characterise our composite part, we inhibited our reporter gene output by adding anhydrotetracycline (AHT), which should bind to TetR. The resulting AHT-TetR complex can no longer repress cI. This should shift the RecA730-cI equilibrium and allow us to observe a deceleration of the flourescence increase. This is consistent with our results. (Figure 7)

In conclusion, we were able to gather data which matched our expectations. Our genetic switch seems to work just as expected and shows little to no basal expression, which is important to avoid the unintentional induction of holin expression. Nevertheless, there are still a lot of experiments to be conducted. Certainly, one of these has to be the induction of phage expression in the lysogen, which we planned to do but could not carry out because of the limitations imposed by the prevailing pandemic.
Biosafety
Inversion via Cre-Lox Recombination
The functionality of our kill-switch is based on the successful and irreversible inversion of a promoter cassette via Cre-Lox recombination. To achieve a visual representation of this inversion, we decided using the two fluorescence reporter proteins superfolder GFP (sfGFP) and mKate2 with additional LVA-ssrA degradation tags. Using fluorescence is a quick and easy way to verify that your gene of interest has been expressed, thus directly confirming promoter activity. Otherwise, methods as Western blot have to be used in order to differ expression levels, requiring the usage of expensive antibodies. We used both reporter genes as they were proven to be functional in B. subtilis.8 As a consequence, we would have used those to test the theoretical constructs for B. subtilis in the lab as well thereby improving comparability regarding the E. coli construct. We used two plasmid backbones for cloning and assembly and thus had to do a co-transformation. The promoter cassette was cloned into the pDest_T7_Cm plasmid with the required T7 polymerase under control of the lac repressor. Hence, the required co-transformation was done using E. coli NEB®10-beta strain in the first place. Since it is an optimized strain for cloning, we expected high transformation efficiency.9 As a result, this assay design enables us to distinguish between the basal T7 promoter driven expression and the IPTG– induced expression rate of sfGFP.
Prior to the inversion, the T7 promoter controls expression of the sfGFP gene. mKate2 should only be expressed at minimal levels in this uninduced state of the switch. This is achieved by with no adjacent promoter region. We should thus only detect a fluorescent signal for sfGFP.
The expression of the cre gene, located on the pJUMP27_araC_pBAD plasmid, is tightly controlled via the araBAD promoter. Upon induction with arabinose, the cassette flanked by the mutant lox sites lox66 and lox71 is inverted via Cre-Lox recombination.10
After induction and subsequent inversion, we expect an increase in mKate2 and a decrease in sfGFP production levels and fluorescence signals due to the degradation tag. The fluorescent signal should thus transition from 510 nm to 615 nm over time. As a result, depleting production of sfGFP and degradation of the remaining reporter are expected to result in basal fluorescence intensity.
We are aware that complete inversion of our cassette upon induction of cre expression is critically important to proper functionality of our switch. Therefore, we plan on employing qPCR for quality control and quantification of inversion efficiency by Cre-Lox recombination. One issue we could face is that the cassette is unintentionally reinverted by Cre recombinase, due to remaining activity towards the lox72 fusion site.10
All in all, if Cre-Lox recombination should prove functional and effective, this proof of concept assay could be the starting point and foundation for further testing in B. subtilis.
References
- 1. Konkol MA, Blair KM, Kearns DB. Plasmid-Encoded ComI Inhibits Competence in the Ancestral 3610 Strain of Bacillus subtilis. Journal of Bacteriology. 2013:4085–4093. http://dx.doi.org/10.1128/JB.00696-13. doi:10.1128/jb.00696-13
- 2. Wu Y, Wang C-W, Wang D, Wei N. A Whole-Cell Biosensor for Point-of-Care Detection of Waterborne Bacterial Pathogens. ACS Synthetic Biology. 2021:333–344. http://dx.doi.org/10.1021/acssynbio.0c00491. doi:10.1021/acssynbio.0c00491
- 3. Saeidi N, Wong CK, Lo T, Nguyen HX, Ling H, Leong SSJ, Poh CL, Chang MW. Engineering microbes to sense and eradicate Pseudomonas aeruginosa , a human pathogen. Molecular Systems Biology. 2011:521. http://dx.doi.org/10.1038/msb.2011.55. doi:10.1038/msb.2011.55
- 4. Lequette Y, Lee J-H, Ledgham F, Lazdunski A, Greenberg EP. A Distinct QscR Regulon in the Pseudomonas aeruginosa Quorum-Sensing Circuit. Journal of Bacteriology. 2006:3365–3370. http://dx.doi.org/10.1128/jb.188.9.3365-3370.2006. doi:10.1128/jb.188.9.3365-3370.2006
- 5. Venturi V. Regulation of quorum sensing inPseudomonas. FEMS Microbiology Reviews. 2006:274–291. http://dx.doi.org/10.1111/j.1574-6976.2005.00012.x. doi:10.1111/j.1574-6976.2005.00012.x
- 6. Chapter One: The master elements of control. In: A Genetic Switch: Phage Lambda Revisited. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2004.
- 7. Vlašić I, Šimatović A, Brčić-Kostić K. Genetic Requirements for High Constitutive SOS Expression in recA730 Mutants of Escherichia coli. Journal of Bacteriology. 2011:4643–4651. http://dx.doi.org/10.1128/jb.00368-11. doi:10.1128/jb.00368-11
- 8. Guiziou S, Sauveplane V, Chang H-J, Clerté C, Declerck N, Jules M, Bonnet J. A part toolbox to tune genetic expression inBacillus subtilis. Nucleic Acids Research. 2016:gkw624. http://dx.doi.org/10.1093/nar/gkw624. doi:10.1093/nar/gkw624
- 9. CompCell_Brochure_1018_UK_LR. https://www.neb.uk.com/neb/media/documents/CompCell_Brochure_1018_UK_LR.pdf
- 10. Zhang Z. Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Research. 2002:90e–990. http://dx.doi.org/10.1093/nar/gnf089. doi:10.1093/nar/gnf089