
Graphical Abstract

Biocontainment
Biocontainment Strategy
When synthetic biology went from a theoretical concept into actual applicable systems, a semblance of doubt began to grow within the scientific community: how do I ensure that my genetically modified system remains safe? Thus, the concept of biocontainment was born. Initially, most biocontainment systems were purely mechanical, such as gloves or sterilization, but at some point, the different applications of modified organisms made the implementation of mechanical methods all but impossible. Last year’s iGEM TU Darmstadt Team decided to design a B. subtilis biofilm that could degrade antibiotics in wastewaters.1 Obviously, rogue colonies floating around had to be avoided but mechanical methods were likely to interfere with the function of the biofilm. So, they added a system that would use the signaling molecules within the biofilm to limit the ability of the bacteria to live outside of it, a system that was further developed by us through the conception of new assays to test its efficacy. We call this system our kill-switch.

The kill-switch relies on an interaction within the biofilm called quorum sensing (QS), more specifically, the ComQXPA system.2 ComX is a 4-amino acid long pheromone, originating out of a 52-amino acid prepeptide that is cleaved and modified by the prenyltransferase ComQ.ComX is then released into the extracellular medium, where it is detected by other B. subtilis cells. A sufficient ComX concentration leads to the autophosphorylation of the histidine kinase domain of ComP, which also releases a phosphoryl group. The now released phosphoryl group is transferred to the kinase ComA, which in turn activates the expression of the comS gene. ComS competes with ComK, the central regulator of competence or the so-called K-State, for the binding site of the MecA-ClpC-ClpP protease. This protease is known to degrade ComK when bound thus by taking its place ComS allows ComK to activate more than 100 genes needed for competence.3 Several genes depend on this signaling cascade including degQ, a non-essential pleiotropic regulator. Its synthesis is promoted by ComA, which means that its promoter is indirectly influenced by ComX.4 In our kill-switch design, we want to use the promoter PdegQ to condition the expression of an essential gene. Our chosen gene is rpsB, which codes for a protein of the 30S subunit of the B. subtilis ribosome. By placing PdegQ before rpsB, we would effectively activate the expression of an essential gene only if the cell is part of the biofilm.5 As highlighted in Figure 2, when a single cell leaves the biofilm, the ComX concentration should not be high enough to activate the signaling cascade, leading to cell death.
Bottlenecks
Since our biocontainment strategy relies on QS, it is a necessity that this signaling cascade is not affected by environmental interactions of the biofilm with its surroundings. A high degree of orthogonality needs to be guaranteed in order to make the system independent of external factors. The ComQXPA-system is unique to Firmicutes such as Bacillus species.6 Thus, the presence of Gram-negative bacteria with their own QS-signaling7 does not interfere with the QS-system of B. subtilis. But this alone is not sufficient to justify a direct application. As we are testing the system on a laboratory scale, it is isolated from a lot of real-world interferences. Therefore, it is nearly impossible to predict the actual interactions of the biofilm community with its surroundings before implementation. Even though cell viability is coupled to containment of cells in our system, new assessment methods need to be developed to ensure that lab observation and field prediction are equally as accurate when regarding biocontainment efficacy.8
Chemical biocontainment strategies are widely applied and involve a huge range of cellular mechanisms.9 Nevertheless, a regulation solely based on a QS-controlled genetic circuit is associated with an inherent risk. In biological systems, mutations caused by e.g., error-prone proofreading function or UV-light exposure cannot be prevented to a 100% extent due to fast evolution of bacteria. A real-world application is thus made difficult by the lack of required safety criteria. We should definitely use an extra barrier to prevent GMOs from escaping the biofilm environment due to spontaneous mutations with the potential to render the QS-system inactive. Moreover, if multiple cells detach from the biofilm in form of clusters, QS threshold concentrations might be maintained despite the release of GMOs.
A great approach to overcome this challenge would be to add a physical biocontainment strategy as an extended safety layer. This could be achieved for instance via an encapsulation by a hydrogel matrix. Within the hydrogel-core, functional biofilms would have a certain degree of mechanical protection.10
Doing so, several conditions need to be fulfilled because the hydrogel-coating would indeed affect the whole project. The pore size of the hydrogel must enable the diffusion of QS-molecules, originating from threatening pathogens, as well as inducers activating genetic circuits. The bacteriophages, as part of the immune-like response, must also penetrate the pores to reach their target.
Most importantly, cell-cell-communication between Bacillus co-cultures as well as optimal growth and accessibility of essential metabolites must not be hindered to guarantee cell viability.
If all the requirements above can be fulfilled within the hydrogel-matrix, the combination of both strategies would be an effective tool to enable a future real-world application of our system within functional biofilms. This would open up PHIRE BYRD to a wide application range for various projects related to biofilm platforms.
Kill-Switch
General Concept
A kill-switch is a mechanism which allows to induce cell death with a certain trigger. It is a common safety strategy that prevents GMOs from getting outside of a controlled test area into the environment. For more information on the importance of biocontainment take a look at our Biocontainment Strategy.
A frequent way to implement a kill-switch is using the concept of auxotrophy by making the organism chemically dependent on a key molecule. Auxotrophic organisms cannot synthesize certain compounds needed for their survival or reproduction.11 There are multiple options for achieving such dependency.
A simple way is to tie an essential part of the metabolism to the presence of a certain compound provided in the lab e.g., a synthetic amino acid. The “Passcode” switch works similar but is a little more complex: The survival of the cell depends on multiple substances rather than one. Only a cocktail of those substances in the appropriate concentration keeps the metabolism running.12 It is literally like a passcode that you must enter to access your bank account: All digits must be correct. This protects your account and is much safer than a code with only one digit.
Another approach would be the “Deadman” switch. Here, the kill-switch cassette encodes a toxin that, when expressed, will rapidly kill the cell from within. However, this can be prevented by an antitoxin: It blocks the transcription of the toxin gene, keeping the cell alive – but only in an antitoxin-supplemented medium. Conversely, this means that the cells die in medium without antitoxin.12
A recent biocontainment strategy is the minimal genome approach. Many genes have situational importance to organisms and are beneficial under selection pressure but are not essential. By reducing the genome to the bare minimum needed for survival under lab conditions, survival in nature becomes very unlikely.11
Whenever an organism is genetically modified, including a kill-switch should be considered to increase biosafety. But there always remains a small chance that the organism still survives. It should be noted that chemical biocontainment strategies are never a 100% reliable (see Bottlenecks).
Design
To ensure biocontainment in our project and prevent the GMOs from escaping into the environment, we further developed the kill-switch from last year’s iGEM Team of TU Darmstadt.1 The kill-switch is based on a genetic circuit that requires the presence of ComX, a quorum sensing molecule, for the expression of an essential gene. We chose the rpsB gene for this purpose as it encodes a protein of the 30S subunit of the ribosome.5 Without functional ribosomes, the cell cannot synthesize proteins and will die. By controlling rpsB expression, we can not only control the reproduction of the cell, but also its’ survival.
ComX usually is only present in Bacillus subtilis biofilm communities. It is required for a signaling cascade resulting in the phosphorylation of ComA to ComA-P. Phosphorylated ComA acts as an inducer of the PdegQ promoter. In our design, this promoter regulates the expression of rpsB, while the native rpsB gene was knocked out previously. In low concentrations of ComX and absence of ComA-P respectively, this promoter cannot be induced. Thus, rpsB will not be expressed, leading to the death of the cell as soon as it leaves the biofilm.
However, during the growth phase of the biofilm, ComA-P concentration might not be high enough to induce the expression of rpsB, resulting in cell death or great reduction of growth. Therefore, we integrated the constitutive promoter Pveg into our promoter cassette. This ensures a constant expression of rpsB during the growth phase, ensuring survival of the cells. Once the biofilm has matured and the concentration of ComA-P is high enough to maintain cell survival, the constitutive promoter is replaced by the inducible promoter PdegQ. But how can we switch between a constitutive and a quorum sensing promoter?
This is where the Cre-Lox system comes in: This recombination system makes it possible to delete or invert a specific DNA sequence via the enzyme Cre recombinase. Two recognition sites, the so called lox sites, flank the sequence of interest and depending on the orientation of the lox sites Cre recombinase either deletes or inverts the sequence.13
In our case, we wanted the Cre recombinase to invert the promoter cassette upstream of the essential gene rpsB – thereby replacing the constitutive promoter Pveg with the quorum sensing promoter PdegQ. We asked Prof. Dr. Lutz for some feedback and got some helpful advice on the correct orientation of the lox sites and our construct. We decided to use lox66 upstream and lox71 downstream of the promoter cassette. Those are mutated, directional recognition sites that allow the irreversible inversion of the flanked DNA sequence when pointing towards each other.

For activating the kill-switch, the expression of the cre gene needs to be inducible. Therefore, we used the xylose-inducible promoter Pxyl from B. subtilis to regulate cre expression. This makes it possible for us to “turn on” the kill-switch by simply adding xylose (Figure 3).
First, rpsB is expressed constitutively by Pveg. Once the cell density and therefore the concentration of ComX is sufficient to ensure cell survival, we add xylose to the biofilm. The expression of cre is induced resulting in an inversion of the promoter cassette by synthesized Cre recombinase. Now, the expression of rpsB is solely regulated by PdegQ and so indirectly by a high cell density within the biofilm and the presence of ComX (Animation 1).
In the presence of ComX, the quorum sensing cascade is initiated: ComA is phosphorylated to ComA-P, which triggers PdegQ and therefore the expression of rpsB. The cell’s ribosomes are functional and the cell survives (Animation 2).
As soon as a cell or a cluster of cells leaves the biofilm, the concentration of ComX drops and PdegQ activation cannot be sustained. Since the inversion of the promoter cassette should be irreversible, rpsB is not expressed anymore, the 30S unit of ribosomes is not renewed within the cell and the translation machinery of the cell will come to a halt, resulting in cell death (Animation 3).
Logic Gate
During the development of our kill-switch, we saw a link between our system and a computational logic gate, which is a model to represent a Boolean operation with one or more binary inputs that lead to a single binary output. The idea of building this type of circuit using biological components is not new.14 We met with Dr. Thomas Gorochowski, an expert in this field, who helped us to translate our design into a logical circuit (Figure 4) that looks as follows:

Figure 4. Kill-switch represented as logic gate. The triangle symbol represents a NOT-Gate. ComX and xylose act as inputs, the desired output is cell death. The AND-Gate only gives an output (1) if both inputs are 1.
Our two inputs are the QS-molecule ComX and xylose. The role of latter in our kill-switch has already been explained (see Design). The desired output is the death of the modified B. subtilis cell if it escapes from the biofilm. Taking all of this into account we made a so-called truth table where we tested the outcomes of all possible combinations:

As we can see in Figure 5 above, only when ComX is absent (0) and xylose has been added to the culture (1) the cell dies (1). This should only happen if the cell leaves the biofilm environment, thus avoiding the release of genetically modified material into the surroundings.
Given the condition that we could only use E. coli to conduct our experiments, we decided to test the efficacy of our cassette design by replacing the essential gene with a fluorescent protein. As this experiment only tests the inversion of our construct and relies on the constitutive promoter T7 for induction, the binary outcomes look different, as seen in the following figure:

The desired output (Figure 6) should be a strong expression of the mKate2 gene after arabinose and IPTG induction (1), given that the Cre-Lox system in combination with the constitutive promoter functions as an AND-Gate.
The biggest challenge in representing our circuit as a logic gate is the fact that it is not a perfect Boolean system. Signaling pathways are always subject to fluctuations and leakiness and are mostly dependant on specific intermolecular interactions. In this case, the expression of rpsB should stop after the ComX concentration falls under a certain level, but it does not need to disappear completely. Besides, after xylose is added, an irreversible change in the structure of the gene occurs, thus rendering the influence of the xylose concentration irrelevant beyond this point. Nevertheless, the similarities with an actual computer system are big enough to allow our system to possibly be optimized using resources from the computer sciences.
Assay Designs
Theoretical Elaboration: B. subtilis
We have designed the following five assays to test our kill-switch in B. subtilis. Due to restricted lab time, however, we were not able to test these assays and they remain theoretical in nature. The assay we were able to conduct is the one optimized for work in E. coli.
There are a few key differences between our B. subtilis and E. coli assay designs. Most notably, the bidirectional promoter cassette (Figure 7) staying true to the original kill-switch design contrary to the unidirectional T7 promoter used in the E. coli proof of concept design.
We initially planned to use GFP and mCherry as reporter proteins, but later settled on sfGFP and mKate2 (for further information see Practical Application: E. coli). Moreover, as the PdegQ promoter3 is part of the cassette to be tested in B. subtilis, we considered using the respective degQ terminator for transcription termination of our coding sequences. However, the degQ terminator would have also been subsequently used with the Pveg promoter. Based on the suggestion of Prof. Dr. Veening, we then decided to use the rrnB terminator15, as it is a strong and universally available terminator, to terminate each coding sequence in our B. subtilis assay designs.
Assay 1: Basal Expression of Pveg
The 1st assay was designed to test the basal expression of the constitutive promoter Pveg16 (Figure 7). Here, the promoter cassette contains the two promoters, Pveg and PdegQ, as well as the two lox sites, lox 66 and lox 71. Both promoters are flanked by two fluorescent reporter proteins: GFP is positioned downstream of Pveg and mCherry upstream of PdegQ.

In this initial state, only the constitutive promoter Pveg should be active and enable the expression of gfp. Hence, GFP fluorescence should be measured with a plate reader at excitation and emission wavelengths of 395 nm and 510 nm17 respectively. Since there is no Cre recombinase nor xylose included in this assay, the construct will not be inverted, and the promoter cassette remains in its initial state. Therefore, GFP should be continuously synthesized and degraded, demonstrating the constitutive function of Pveg. As there is no ComX included either, the quorum sensing-dependent promoter PdegQ should not be activated and thus no mCherry should be produced. Hence, plate reader measurements can be conducted to assess the leakiness of PdegQ, demonstrated by mCherry fluorescence. In that case, mCherry should be measured at excitation and emission wavelengths of 587 nm and 610 nm18 respectively. The results could be used as calibration values for the upcoming assays.
Assay 2: Activation of PdegQ using a ComX Gradient
In the 2nd assay we want to identify the optimal concentration of ComX needed to indirectly activate the QS-dependent promoter PdegQ. Via the two-component system (TCS) of B. subtilis, the quorum sensing molecule ComX initiates a reaction cascade, leading to induction of PdegQ by ComA-P (as explained in Biocontainment Strategy).
For this purpose, the same construct as seen in Figure 7 can be used. Starting in the nanomolar-concentration range19, we would add ComX to the respective wells in our well plate following an ascending gradient. If the concentration of ComX surpasses the activation threshold, we should detect a fluorescent signal for mCherry. This should allow us to determine the optimal ComX concentration for the induction of the mCherry synthesis. We would use that concentration as a starting point for testing the expression of the essential gene rpsB in Assay 4.
Assay 3: Inversion via Cre-Lox Recombination
The 3rd assay examines the inversion capacity of the promoter cassette by Cre-Lox recombination. The construct we have devised in Figure 8 is similar to the previous one (Figure 7) but additionally contains a cre gene regulated by PXylA, a xylose-inducible promoter.20

Analogous to Assay 1, no ComX is added and consequently, only Pveg should display activity. Therefore, only GFP should be produced. Once xylose is added, Cre monomers will be synthesized and form two dimers, attaching to the palindromic sequences of each lox site.21 Subsequently, the promoter cassette will be inverted and Pveg should now regulate the expression of mcherry. The successful inversion by Cre recombinase can thus be detected with a plate reader as a shift from green to red fluorescence.
Assay 4: Cell Viability
The 4th assay was designed to determine cell viability while PdegQ is regulating gene expression of the essential gene rpsB.22 As depicted in Figure 9, the promoter cassette is already inverted in this construct. Here, PdegQ is regulating rpsB expression. This was done to minimize any potential error sources.

The optimal ComX concentration, previously determined in Assay 2, is now used as a starting point to test the expression rate of rpsB. The native rpsB gene in our B. subtilis strain will be knocked out and replaced by our construct. ComX is then added in ascending concentrations to the cultures. Individual cells should therefore only be able to survive if the ComX concentration reaches the threshold to indirectly induce the expression of rpsB to a sufficient degree.
The determined threshold of ComX should be surpassed within a biofilm community and not reached outside of the biofilm, thus leading to cell death after escape.
Cell survival is assessed using Live/Dead staining of which we have found many different approaches.23,24 One very simple, exemplary method works by using the dye trypan blue to stain a sample of cells. Live cells do not take up the dye and appear colorless. Dead cells with damaged membranes cannot prevent the dye from entering – hence, they will be stained blue.25 The respective number of cells can be determined with a hemocytometer which lets us count the cells using a microscopic slide with a gridded coverslip.
Assay 5: Inversion Efficiency
Lastly, the 5th assay evaluates the efficiency of the Cre-mediated inversion of our kill-switch cassette. To ensure biocontainment within our whole project design, the inversion rate of the promoter cassette needs to be nearly if not a 100%. Our previous assays have been qualitative in nature but for this assay, we needed a quantitative analysis of our kill-switch. We first considered using a Southern blot but were advised by Prof. Dr. Lutz to conduct a qPCR analysis.
Identical to a regular PCR, primers are needed to amplify sequences of interest – in our case the inverted promoter cassette. The primers we chose would be complementary to the junctions of the mutated loxP/lox72 sites and the inverted promoters. As such the promoter cassette would only be amplified once the inversion has taken place.26
One way of implementing such an analysis is using fluorophores, such as SYBR green, which absorbs light at 485 nm and emits green fluorescent light at 524 nm.27 It binds to DNA, specifically the minor groove of double-stranded DNA, and fluoresces in this bound state. While it can bind to single-stranded DNA during the annealing stages of the qPCR cycles, SYBR green only produces a low fluorescence in that state. This low fluorescence can be used as calibration for background emissions. Consequently, SYBR green would only reach threshold concentrations beyond the background noise once the inverted cassette is amplified. The amplified cassette forms a double helix for the dye to intercalate and fluoresce, thus demonstrating a successful inversion of our kill-switch cassette.26
Practical considerations regarding the qPCR included positive and negative controls. Among the positive controls is a dilution series of the inverted construct, which would represent a 100% amplification. The negative control would be a blank with merely water or buffer, as well as a No-Template control with the non-transformed B. subtilis strain, thus excluding amplification events. Three concentrated samples of the inverted construct would be tested via qPCR at defined timepoints after induction of cre expression and thus Cre-Lox recombination.
To interpret the data, we would employ the Δ-Δ Ct method28 using the controls as reference to determine the relative changes in quantity of our inverted construct. We would also perform a gel electrophoresis to evaluate the identity of the amplification products and confirm a successful qPCR.
Practical Application: E. coli
Adaptation of Parts
To validate our proof of concept assay for the kill-switch, we decided to work in E. coli. E. coli is a well-studied model organism and unlike B. subtilis, was already established in our laboratory. Hence, we could use approved lab protocols. Additionally, due to the fact that molecular cloning and transformation of B. subtilis are far more challenging than for E. coli29, this step was beneficial to the upcoming lab work, enabling us to run more experiments. We could thus obtain more representative data through better statistics. We kept Golden Gate Assembly as cloning strategy, so all parts were designed to be flanked by BsaI recognition sites. Thus, all basic parts were optimized for the work in E. coli.

Initially, we codon-optimized the sequences (CDS) for sfGFP, mKate2 and cre for efficient translation in E. coli. We then introduced the ribosomal binding sites30 (RBS), while making sure that at least four nucleotides as core motif of the Shine-Dalgarno consensus sequence 5’‑GGAGG‑3′ were present.31,32 Furthermore, we replaced the constitutive Pveg promoter and rrnB terminator with the well-characterized T7 promoter and T7 terminator of E. coli respectively. Additionally, we adapted the sequence of the LVA-ssrA degradation tag for use in E. coli, corresponding to the sequence (A)ANDENYALAA**.33
As mentioned in the section Theoretical Elaboration: B. subtilis previously, the kill-switch cassette to be tested would be based on a bidirectional promoter cassette with Pveg and PdegQ pointing in opposite directionalities as constitutive and inducible promoter respectively. As the ComQXPA-gene cluster is not present in Gram-negative bacteria6, we opted for an alternative inducible promoter to exchange PdegQ. To maintain tight regulation of the circuit upon induction, while adapting to a new chassis, choosing the right promoter to replace PdegQ was critical. One consideration was to leave PdegQ as a placeholder. This idea was quickly discarded, as potential background activity of PdegQ caused by an unknown trigger could not be excluded. Consequently, we decided against the usage of a bidirectional promoter cassette to reduce the assay complexity. As a result, only the T7 promoter controls gene expression and is affected by inversion (Figure 10).
Narrowing down the complexity to a minimum, the design of our proof of concept assay is easily adaptable to be applied in other experimental setups. Other research groups or future iGEM Teams could further use this assay to engineer translation control by shuffling of the RBS to tailor-fit the system to their switch. Furthermore, when replacing the promoter, evaluation of promoter strength could also be conducted, while remaining with the same CDS to ensure comparability. Since the assay complexity is reduced, troubleshooting should also prove easier compared to the original system.
Proof of Concept
To prove the functionality of our kill-switch and thus confirming Cre-Lox inversion, we developed a proof of concept assay to be tested within E. coli. For further information, visit the Proof of concept page.
DBTL
We thoroughly examined potential weaknesses of our kill-switch cassette and discussed further steps to optimize our kill-switch to be ready for real-world implementation. Therefore, have a look at the Engineering page.
Lab Results
In order to check that we were able to successfully clone the cre gene into the pJUMP27_araC_pBAD backbone via Golden Gate Assembly, we sequenced our plasmid twice. Both sequencing results were aligned to the plasmid map of our construct in SnapGene® as shown in Figure 11.
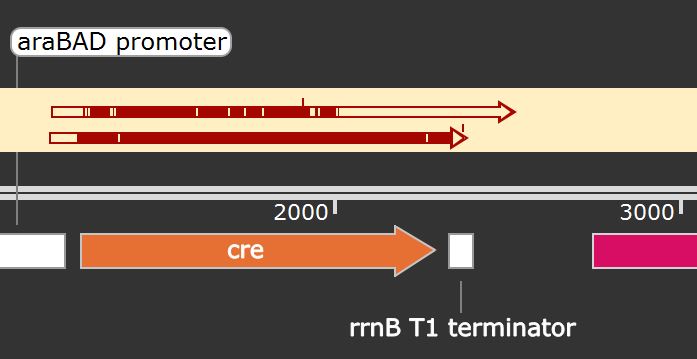
The first sequencing had a lot of point mutations, indicated by the white bars present in the first arrow on top (Figure 11), and was hence repeated from Eurofins.

Our second sequencing result showed a mismatch at sequencing position 205 bp (Figure 12): there is an overlapping peak between A (green) and G (white). Since the guanine was correctly detected within the first sequencing, the plasmid sequence should be correct. This mismatch is inconclusive and can normally occur within Sanger sequencing, especially when sequencing larger constructs. Thus, we can expect that synthesized Cre recombinase should be functional. In conclusion, through both sequencing results, we can confirm that the cloning results were positive.
Unfortunately, we could not obtain any fluorescence data for the proof of concept assay as we had to cut experiments in the laboratory.
References
- 1. TU Darmstadt G. B-TOX: Reduction of wastewater toxicity by using a B. subtilis biofilm. 2020. https://2020.igem.org/Team:TU_Darmstadt
- 2. Špacapan M, Danevčič T, Štefanic P, Porter M, Stanley-Wall NR, Mandic-Mulec I. The ComX Quorum Sensing Peptide of Bacillus subtilis Affects Biofilm Formation Negatively and Sporulation Positively. Microorganisms. 2020;8. doi:10.3390/microorganisms8081131
- 3. Miras M, Dubnau D. A DegU-P and DegQ-Dependent Regulatory Pathway for the K-state in Bacillus subtilis. Frontiers in microbiology. 2016;7:1868. doi:10.3389/fmicb.2016.01868
- 4. Ogura M, Yamaguchi H, Yoshida K, Fujita Y, Tanaka T. DNA microarray analysis of Bacillus subtilis DegU, ComA and PhoP regulons: an approach to comprehensive analysis of B.subtilis two-component regulatory systems. Nucleic acids research. 2001;29:3804–3813. doi:10.1093/nar/29.18.3804
- 5. Zhu B, Stülke J. SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic acids research. 2018;46:D743–D748. doi:10.1093/nar/gkx908
- 6. Dogsa I, Choudhary KS, Marsetic Z, Hudaiberdiev S, Vera R, Pongor S, Mandic-Mulec I. ComQXPA quorum sensing systems may not be unique to Bacillus subtilis: a census in prokaryotic genomes. PloS one. 2014;9:e96122. doi:10.1371/journal.pone.0096122
- 7. Miller MB, Bassler BL. Quorum sensing in bacteria. Annual review of microbiology. 2001;55:165–199. doi:10.1146/annurev.micro.55.1.165
- 8. Developing Microbial Biocontainment Strategies and Their Assessment Methods | Research Project Database | Grantee Research Project | ORD | US EPA. 9AD.
- 9. Lee JW, Chan CTY, Slomovic S, Collins JJ. Next-generation biocontainment systems for engineered organisms. Nature chemical biology. 2018;14:530–537. doi:10.1038/s41589-018-0056-x
- 10. Tang T-C, Tham E, Liu X, Yehl K, Rovner AJ, Yuk H, de La Fuente-Nunez C, Isaacs FJ, Zhao X, Lu TK. Hydrogel-based biocontainment of bacteria for continuous sensing and computation. Nature chemical biology. 2021;17:724–731. doi:10.1038/s41589-021-00779-6.
- 11. Whitford CM, Dymek S, Kerkhoff D, März C, Schmidt O, Edich M, Droste J, Pucker B, Rückert C, Kalinowski J. Auxotrophy to Xeno-DNA: an exploration of combinatorial mechanisms for a high-fidelity biosafety system for synthetic biology applications. Journal of Biological Engineering. 2018. http://dx.doi.org/10.1186/s13036-018-0105-8. doi:10.1186/s13036-018-0105-8
- 12. Chan CTY, Lee JW, Cameron DE, Bashor CJ, Collins JJ. “Deadman” and “Passcode” microbial kill switches for bacterial containment. Nature Chemical Biology. 2015:82–86. http://dx.doi.org/10.1038/nchembio.1979. doi:10.1038/nchembio.1979
- 13. Zhang Z, Lutz B. Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic acids research. 2002;30:e90. doi:10.1093/nar/gnf089.
- 14. Bartoli V, Meaker GA, Di Bernardo M, Gorochowski TE. Tunable genetic devices through simultaneous control of transcription and translation. Nature communications. 2020;11:2095. doi:10.1038/s41467-020-15653-7
- 15. Krásný L, Gourse RL. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. The EMBO journal. 2004;23:4473–4483. doi:10.1038/sj.emboj.7600423
- 16. Guiziou S, Sauveplane V, Chang H-J, Clerté C, Declerck N, Jules M, Bonnet J. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic acids research. 2016;44:7495–7508. doi:10.1093/nar/gkw624.
- 17. Remington SJ. Green fluorescent protein: a perspective. Protein science : a publication of the Protein Society. 2011;20:1509–1519. doi:10.1002/pro.684
- 18. Cloin BMC, de Zitter E, Salas D, Gielen V, Folkers GE, Mikhaylova M, Bergeler M, Krajnik B, Harvey J, Hoogenraad CC, et al. Efficient switching of mCherry fluorescence using chemical caging. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:7013–7018. doi:10.1073/pnas.1617280114
- 19. Dogsa I, Spacapan M, Dragoš A, Danevčič T, Pandur Z, Mandic-Mulec I. Peptide signaling without feedback in signal production operates as a true quorum sensing communication system in Bacillus subtilis. Communications biology. 2021;4:58. doi:10.1038/s42003-020-01553-5
- 20. Rygus T, Scheler A, Allmansberger R, Hillen W. Molecular cloning, structure, promoters and regulatory elements for transcription of the Bacillus megaterium encoded regulon for xylose utilization. Archives of microbiology. 1991;155:535–542. doi:10.1007/BF00245346
- 21. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis (New York, N.Y. : 2000). 2000;26:99–109.
- 22. Lauber MA, Running WE, Reilly JP. B. subtilis ribosomal proteins: structural homology and post-translational modifications. Journal of proteome research. 2009;8:4193–4206. doi:10.1021/pr801114k
- 23. Boulos L, Prévost M, Barbeau B, Coallier J, Desjardins R. LIVE/DEAD BacLight: application of a new rapid staining method for direct enumeration of viable and total bacteria in drinking water. Journal of Microbiological Methods. 1999;37:77–86. doi:10.1016/S0167-7012(99)00048-2
- 24. Rosenberg M, Azevedo NF, Ivask A. Propidium iodide staining underestimates viability of adherent bacterial cells. Scientific reports. 2019;9:6483. doi:10.1038/s41598-019-42906-3
- 25. Felice DL, Sun J, Liu RH. A modified methylene blue assay for accurate cell counting. Journal of Functional Foods. 2009;1:109–118. doi:10.1016/j.jff.2008.09.014
- 26. Zhang Q, Wang J, Deng F, Yan Z, Xia Y, Wang Z, Ye J, Deng Y, Zhang Z, Qiao M, et al. TqPCR: A Touchdown qPCR Assay with Significantly Improved Detection Sensitivity and Amplification Efficiency of SYBR Green qPCR. PloS one. 2015;10:e0132666. doi:10.1371/journal.pone.0132666
- 27. Zipper H, Brunner H, Bernhagen J, Vitzthum F. Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications. Nucleic acids research. 2004;32:e103. doi:10.1093/nar/gnh101
- 28. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.). 2001;25:402–408. doi:10.1006/meth.2001.1262
- 29. Zhang X-Z, You C, Zhang Y-HP. Transformation of Bacillus subtilis. Methods in Molecular Biology. 2014:95–101. http://dx.doi.org/10.1007/978-1-4939-0554-6_7. doi:10.1007/978-1-4939-0554-6_7
- 30. Wu Y, Wang C-W, Wang D, Wei N. A Whole-Cell Biosensor for Point-of-Care Detection of Waterborne Bacterial Pathogens. ACS synthetic biology. 2021;10:333–344. doi:10.1021/acssynbio.0c00491
- 31. Saito K, Green R, Buskirk AR. Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. eLife. 2020;9. doi:10.7554/eLife.55002
- 32. Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. Journal of bacteriology. 2002;184:5733–5745. doi:10.1128/JB.184.20.5733-5745.2002
- 33. Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nature structural biology. 2000;7:449–455. doi:10.1038/75843