Previous studies have
suggested that ribosomes can protect mRNA transcripts from RNase activity, possibly by covering
RNase binding sites and limiting their accessibility[5][6].
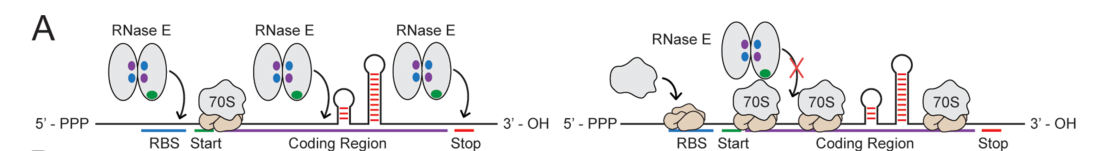
Fig 2
Schematics illustrating how a mRNA’s translation rate controls the accessibility of RNase binding
sites in an operon’s coding regions.[1]
In order to quantify
the relationship between the translation rate and the decomposition rate of mRNA, we constructed a
relevant model to simulate the process.
The key to modeling
is to calculate how many bases are not covered by ribosomes and are exposed to the catalytic range
of the RNase. Here, we designated α as the ratio of the ribosome’s translation initiation rate over
its elongation rate over its elongation rate. We also designated F as the physical footprint of each
ribosome in units of trinucleotides (amino acids). According to a TASEP (totally asymmetric
exclusion process) model of ribosome dynamics that includes the ribosome’s footprint on the
mRNA[7].
When translation
initiation is the rate-limiting step (α is less than one), the steady-state ribosome density ρr
is
$$
\rho _{\mathrm{r}}=\min \left( \frac{F\alpha}{1+(F-1)\alpha},1 \right) \tag{a}
$$
According to the
definition,
$$
\rho r=\frac{\left( MF \right)}{N}
$$
(M is the number of
ribosomes on RNA, and N is the number of codons on RNA)
The relationship
defined by eq illustrates how increasing a mRNA’s translation initiation rate results in higher
ribosome densities, up until the maximum possible value.
Next, we define the
unprotected distance between two adjacent ribosomes, the number of trinucleotides between the end of
one ribosome and the beginning of the next ribosome. We call this unprotected RNA ‘hole’, so the
hole density ρh=1-ρr. According to the TASEP model,the probability that a bound ribosome has m free
trinucleotides.
$$
P(D=m)\propto \frac{\rho _{\mathrm{r}}}{\rho _{\mathrm{r}}+\rho _{\mathrm{h}}}\left( \frac{\rho
_{\mathrm{h}}}{\rho _{\mathrm{r}}+\rho _{\mathrm{h}}} \right) ^m\tag{b}
$$
The first half of the
equation calculates the probability that a mRNA position is bound by a ribosome, whereas the second
part calculates the probability that the next m adjacent positions on the mRNA all contain a hole,
Substituting equation (a) and the definition of the hole density into equation (b), we get the
probability distribution of the distance between ribosomes
$$
P(D=m)\propto \frac{\rho _{\mathrm{r}}\left( 1-F\rho _{\mathrm{t}} \right) ^m}{\left( 1+\rho
_{\mathrm{r}}-F\rho _{\mathrm{r}} \right) ^{(m+1)}}\tag{c}
$$
We then determined
the ribosomes’ average headway distance by calculating the first moment of the probability
distribution in eq(c), using
$$
\langle D\rangle =\frac{1}{Z}\sum_{m=1}^{m=L}{P}(D=m)m\tag{d}
$$
Where $
Z=\sum\nolimits_{m=1}^{m=L}{P(D=m)}
$
According to the
model, we model the following results
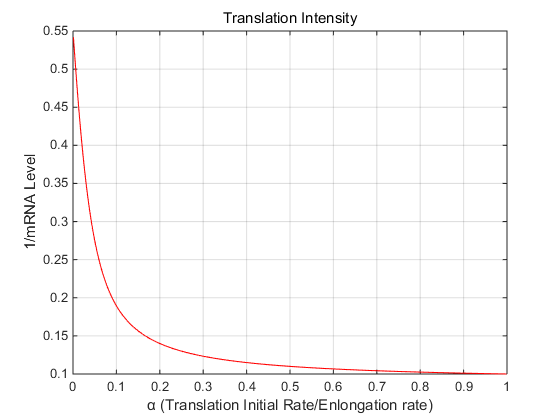
Fig 3 the
translation initial rate affects the rate of RNA decomposition(1/mRNA level)
It can be seen that
with the increase of translation intensity, the rate of RNA decomposition continues to decrease,
verifying the hypothesis proposed in the literature[1].