Team:British Columbia/Experiments
EXPERIMENTS
Experimental Design
INTRODUCTION
Based on our Project Design decisions, we set out to experimentally validate our project’s four main components:
- TNFa-induction
- Lactate-induction
- Split-lux operon AND-gate system
- Inducible promoter screening
CIRCUIT CONSTRUCTION
Construction of psicA Characterization Circuit
The sicA promoter regulates the expression of secreted effort proteins such as Salmonella Invasion Protein (Sip) A in Salmonella (Temme et al, 2008). Ma et al (2010) reported that upon the induction of human TNFa, there was a significant increase of SipA protein production. Hence we hypothesized that sicA was involved in a regulatory pathway in Salmonella that was responsive to TNFa. Since TNFa was identified as a potential immune marker of interest in tumour microenvironments, we decided to further characterize its inductive function to the psicA promoter as a proof-of-concept. Following this proof-of-concept, this validated inducible system would be used to produce a reporter in the presence of both TNFa and our second biomarker, lactate.
To test said inductive function, we are using a reporter-based assay system where psicA is used as a promoter to induce the transcription of a downstream red fluorescent protein gene (RFP). Characterization of pSicA involved harnessing TNFα as a signal to induce expression of the reporter and (Lou et al., 2019). The set-up involves fluorescence quantification of the TNFα inducible pSicA promoter system using a plate reader. We hypothesized that with increasing TNFα concentrations, we would expect to see a dose-dependent upregulation response of our engineered psicA-RFP construct. This result would corroborate results from Ma et al, 2010, which reported that TNFa upregulated sicA production, hence validating an inducible promoter in S.enterica that would be indicative of TNFa-releasing immune cell presence.
Our goal was to design a plasmid containing the psicA promoter with mRFP1 in a high copy number plasmid, pSB1C3, so that we can use it to transform it into Escherichia coli and S.enterica. To do this, we used the Golden Gate Assembly (GGA) method to assemble our parts into a single plasmid by creating customized overhangs allowing assembly via their complementary overhang sequences (Engler et al., 2008).
Repurposing pSBIC3 BBa_J04450 to create a TNFa-inducible RFP plasmid
We designed forward and reverse primers with overhang sequences to extract our desired part sequence of the pSBIC3 BBa_J04450 plasmid. The pSBIC3 BBa_J04450 plasmid construct contains the Lac operon which we cut out using PCR as described in the “Molecular cloning” section below. We analyzed our primer designs with IDT’s Oligo Analyzer. The calculated self-dimer values were below the recommended maximum delta F value of -9.0kcal/mole.
Amplification of the psicA gblock fragment
We ordered a Golden Gate compatible psicA gblock fragment which contained psicA with overlapping overhang sequences that allowed it to anneal with our repurposed “pSBIC3 BBa_J04450 TNFα inducible RFP plasmid” that contain overhang sequences necessary for proper Golden Gate Assembly.
| Final Golden Gate Assembled psicA-RFP Construct |  |
|---|
Construction of pLactate Characterization Circuit
Salmonella enterica serovar Typhimurium has been shown to have endogenous lactate utilization operons, orthologous to the lldPRD operon in E. coli, that are induced by the presence of L-lactate. Through this operon, S. enterica is able to use L-lactate generated by inflammation as an electron donor during gut colonization (Gillis et. al, 2019). Given S. enterica’s ability to self-regulate with L-lactate, we sought to build a bacterial reporter system from parts of the lactate-regulated operon to target the propensity for lactate enrichment seen in many tumour microenvironments.
While the E. coli lldPRD operon has been well characterized, its homologous operon in S. enterica is poorly understood. Therefore, we needed to be able to characterize our construct containing the lactate regulatory units as a proof of concept before we could integrate it into the main split operon circuit. Similar to our psicA-TNFa system, we would activate the inducible promoter with varying concentrations of the induction molecule - in this case lactate - and measure fluorescence of a downstream RFP.
This system behaves according to the negative regulation of the lactate promoter (pLactate). Since pLactate is a constitutively active promoter, the lldR genes are constantly being expressed. However, the regulatory LldR protein is also expressed in this system, which represses the activity of pLactate. In the presence of lactate, LldR is released from the pLactate promoter, allowing for activation of the promoter. Applied to our promoter proof-of-concept system, pLactate and LldR are expressed upstream of an RFP gene to be able to correlate fluorescence to lactate-dependent promoter activity, hence controlling its expression in the presence of lactate. This construct allows us to perform lactate induction experiments to determine lactate dose-response curves at different lactate concentrations. Since lactate was chosen as our second biomarker for immune activity in the tumour microenvironment as described in on our Project Design page, these assays allow us to validate the part’s efficacy at distinguishing healthy and tumour environments. With the information from the dose-response curves and literature on the different lactate concentrations in healthy tissue vs tumours, these assays allow us to validate the part’s efficacy at distinguishing healthy and tumor environments. With the information from the dose-response curves and literature on the different lactate concentrations in healthy tissue vs tumours, we can establish if our biosensor can detect the range of lactate concentrations present in the TME and output an adequately strong response to be sensed in urine.
Circuit Construction of pLactate Reporter Construct
The construct we wanted to build was:

The pLactate and lldR sequences were obtained from the NCBI Genbank Salmonella enterica full genome sequence (Kroger, et. al, 2020). The lldR sequence was found by searching for the gene name in the database (between 3908879 - 3909655 bp). Because the pLactate sequence was not annotated, we needed to narrow down the sequence to be located between lldP and sadA (the regulatory region between the two genes). By cross referencing with the sequence of the E. coli lactate promoter, we were able to utilize the concept of gene conservation to pinpoint where the S. enterica lactate promoter was in the genome (3906960 - 3907177 bp). Because the pLactate sequence was not annotated, we needed to narrow down the sequence to be located between lldP and sadA (the regulatory region between the two genes). By cross referencing with the sequence of the E. coli lactate promoter, we were able to utilize the concept of gene conservation to pinpoint where the S. enterica lactate promoter was in the genome (3906960 - 3907177 bp).
The rest of the sequences were taken from biobricks. They are as follows: pSB1C3 vector, RFP (BBa_E1010), Terminator (first; BBa_J61102), Terminator (second, BBa_B0015), (synthetic) Constitutive Promoter (BBa_J23101), and RBS (BBa_J61102).
1) Using Benchling, IDT’s Oligochecker, and NCBI’s primer blast, we designed Golden Gate primers to make the parts compatible for assembly. Similarly, pLactate and lldR sequences were modified for Golden Gate Assembly by adding Golden Gate cut sites at the flanks of the sequences.
2) We ordered two gblocks, one with the lactate inducible promoter and RBS, and the other was a construct with the constitutive (synthetic) promoter (BBa_J23101), RBS, lldR, and terminator. Parts were ordered with Golden Gate cut sites already incorporated. Primers were ordered for the PCR modification of RFP and the pSB1C3 backbone.
3) We ordered two gBlocks, one with the lactate inducible promoter and RBS, and the other was a construct with the constitutive (synthetic) promoter (BBa_J23101), RBS, lldR, and terminator. Parts were ordered with Golden Gate cut sites already incorporated. Primers were ordered for the PCR modification of RFP and the pSB1C3 backbone.
Split-lux Operon Circuit Construction
A split operon is a dual-plasmid system that serves as a genetic AND-gate. It is created by “splitting” an operon into different parts–two in our project–with each part encoded on a separate plasmid, typically under the control of a unique promoter. Independently, each part of the split operon is nonfunctional. It is only when both parts of the operon are co-expressed, that the aggregate gene products from the different parts can carry out some function within the biological system.
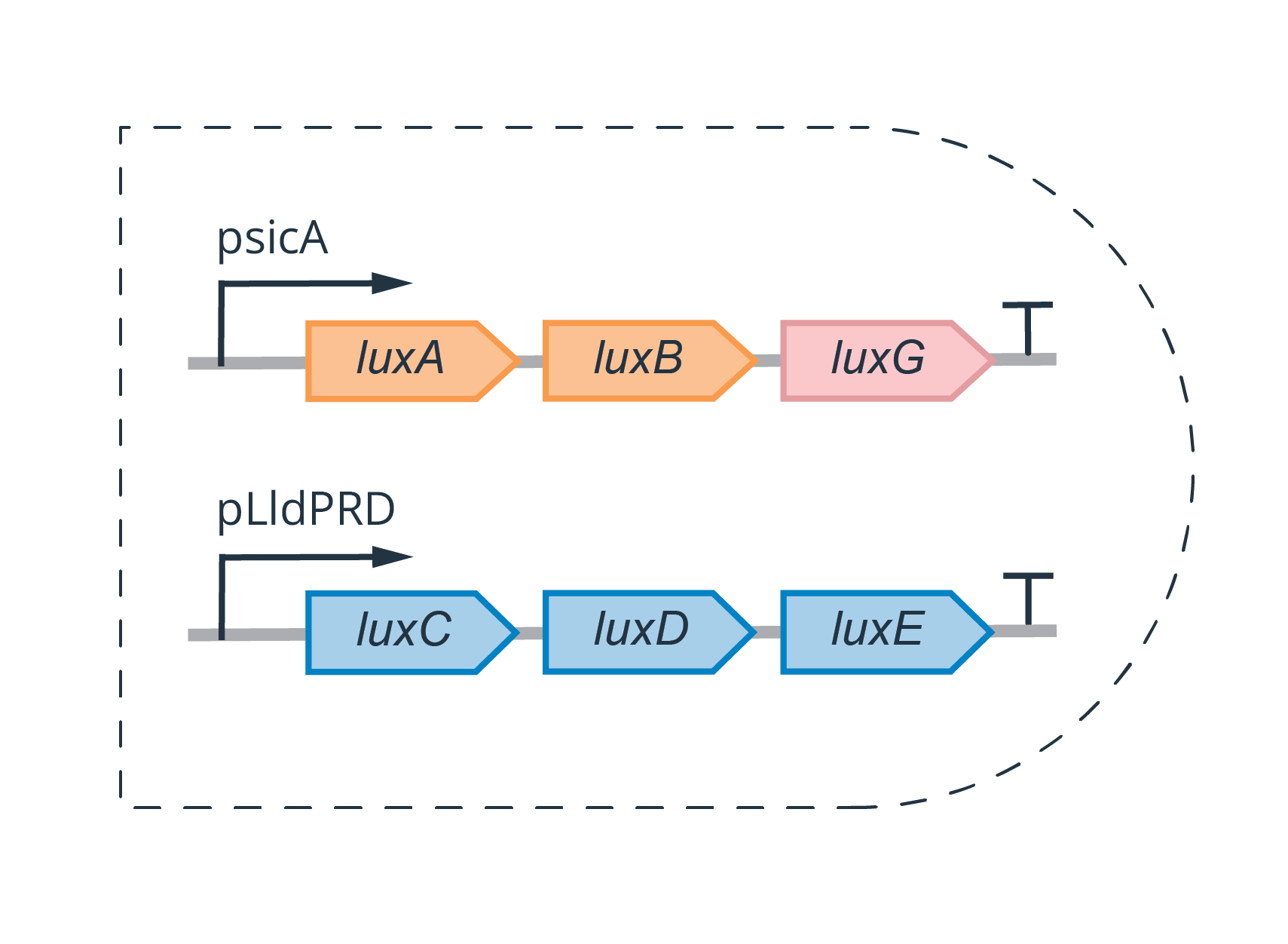
Our project involved splitting the lux operon, which is responsible for bioluminescence in many bacterial families, including Vibrionaceae, Shewanellaceae and Enterobacteriaceae (Brodl et al., 2018). The genes expressed under the lux operon–and responsible for light production–are typically organized in the following order; LuxCDABEG.
For our circuit construction, we designed and ordered gBlocks for each of our insert components, each containing pre-specified overhangs to allow for subsequent Golden Gate reactions. Using these gBlocks, two sets of Golden Gate reactions were carried out, similarly to the workflow described in the section about (psicA circuit construction), which was in accordance with the protocols in our Protocols page.
Circuit 1 was assembled on the pSB1C3 backbone (found in the iGEM registry, and made Golden Gate compatible for BsaI digestion using PCR) — which includes the Chloramphenicol resistance gene.
Circuit 2 was assembled on the pSB1K3 backbone (found in the iGEM registry, and made Golden Gate compatible for BsaI digestion using PCR) — which includes the Kanamycin resistance gene.

Inducible Promoter Screening Vector Construction
Circuit construction for the promoter discovery portion of the wet lab consists of a forward GFP and terminator as well as a reverse RFP and a reverse terminator assembled as shown below. The blunt end BamHI cut site in the middle will serves as the location of insertion of sheared Salmonella genomic DNA fragments.
1) The functional DNA parts required to construct the screening vector could all be found in the iGEM distribution kit, hence we designed PCR primers to amplify the necessary parts to later be assembled via Golden Gate.

2) Molecular cloning would be carried out as described above in the psicA circuit construction section above.
3) Preparation of the screening construct for insertion of sheared Salmonella genome fragments involves production of blunt end fragments following a 2 hour 37 C digestion with BamHI. To blunt sticky ends after BamHI digestion, we would incubate the construct with T4 DNA polymerase and dNTPs for 30 minutes at 11 C as described in Bumann & Balvidia 2007. CIAP treatment after blunting would be used to reduce self-ligation. Resulting DNA fragments would then be used as described in our circuit characterization section for screening for other inducible promoters.
CIRCUIT VALIDATION
psicA characterization
A construct was set-up to characterize the psicA bioinformatically to elaborate induction of the psicA promoter with TNFa, the protocol of the experimental set-up was adapted from Ma et al. 2010: shown below. Due to time constraints, a proof-of-concept experimental procedure could not be carried out, but the protocol is described:
Day before the experiment was run:
1) Salmonella cells with constitutively expressed RFP cells (positive control), cells with an empty plasmid (negative control), and cells with our psicA-RFP construct were inoculated at 37C overnight.
2) Non-agitated microaerophilic bacterial cultures will be prepared by inoculation of 5 ml of broth with 0.01ml of a stationary phase culture with varying concentrations of TNFa to form a dose response curve (i.e. Diluting TNFa solutions to 0ng/mL, 2ng/mL, 4ng/mL, 6ng/mL 8ng/mL, and 10ng/mL), followed by an overnight incubation (~18 h) at 37°C.
Day of the experiment:
1) Overnight cultures of bacteria will be concentrated 33-fold in Hank’s balanced salt solution (HBSS) supplemented with 10 mM HEPES, pH 7.4. The overnight cultures from the TNFa pretreated Salmonella strains will be washed thoroughly with HBSS three times to get rid of potential TNFa residue in the media. The bacteria will then be resuspended in fresh HBSS for cell lysis.
2) Next, cell-lysed bacteria will be loaded onto a 96 well plate using a multichannel pipettor, and fluorescence readout will be measured using plate reader.
Data would then be analyzed and graphed as was done for lactate induction experiments (see below and in our Results page). If the promoter would work as expected, we would observe increasing levels of RFP expression as TNFa concentration increases.
pLactate characterization
As the lactate inducible promoter gets induced once lactate binds to its substrate, we measured the promoter's activity by adding different concentrations of lactate in a plate reader (Goers et al, 2017)
Day before the experiment was run:
1) Salmonella cells with constitutively expressed RFP cells (positive control), cells with an empty plasmid (negative control), and cells with our pLactate-LldR-RFP construct were inoculated at 37C overnight.
2) We prepared the lactate media the day prior. Lactate solutions were diluted to the appropriate concentrations to be put in the reader (0mM, 2mM, 5mM, 15mM, 40mM and 100mM lactate). These concentrations were determined through literature searches of lactate presence in healthy and tumourous human tissue.
Day of the experiment
1) The OD600 of the cells was diluted to 0.5 OD600 (and checked using the spectrometer) so that the concentration of the cells is correct after being added into the media by the liquid dispenser.
2) The lactate media with cells was loaded into a 96 plate using a multichannel pipettor, and fluorescence readout measured using plate reader.
From this assay we hoped to validate the promoter’s nature, as it is induced by different lactate concentrations. If the promoter would be working as expected, we would be able to see a greater RFP expression as lactate concentration increases, and consequently prove that the promoter worked. After running the experiment and collecting the data, we were able to see the promoter getting induced by seeing increasing levels of RFP being expressed as concentration of lactate increased, successfully achieving a proof-of-concept experimental results. To see the figures and explanation of results from this assay, please head over to our Results page or Engineering Success page.
Functional Validation of the Split Operon
In our future experiments, we are planning to validate the functionality of our split operon. After the simultaneous electroporation of the lactate and TNFa inducible plasmids with the split operon to Salmonella, the constructs would be plated on a LB medium plate with chloramphenicol and kanamycin antibiotics. This will ensure that only the bacteria which took up both of the plasmids are growing on the plate. The colonies would be picked the next day and genotyping PCR would be performed to make sure the construct is present. Successful colonies would be inoculated and confirmed once again for successful transfection with restriction digest check with EcoRI and PstI. Upon confirmation of the successful transfection, glycerol stock would be prepared for future use of the constructs.
Next, the bacterial cells will be subjected to various treatments and luciferase light output will be measured to confirm the functionality of the both circuits. After the pSicA-RFP and pLactate-RFP characterization experiments, the optimal concentration for promoter induction will be elucidated. This optimal concentration will be used to induce simultaneous promoter induction in Salmonella with both plasmids present. The following conditions will be tested:
- Salmonella transfected with pSicA-LuxABG and pLactate-LuxCDE with optimal concentrations of lactate and TNFa.
- Salmonella transfected with pSicA-LuxABG and pLactate-LuxCDE with optimal concentrations of lactate and no TNFa.
- Salmonella transfected with pSicA-LuxABG and pLactate-with no lactate and optimal concentrations of TNFa.
- Salmonella transfected with split LuxABG and LuxABG plasmids under the control of the constitutive promoters (Positive Control).
- Salmonella transfected with empty pSB1C3 and SB1K3 backbones (Negative Control).
Concentrations will be changed to lower or higher than optimal concentrations as needed, if no or little luciferase output is detected in the plate reader. Experiments will be performed in triplicates to ensure statistical significance.
Given that the system that is relevant for implementation of this model is human, we would like to explore validation in hydrogel tumor microenvironment (TME) reconstructions. However, beyond this, another important avenue we would like to explore is hydrogel tumour microenvironment (TME) reconstructions. 2D tumour models, like cell lines, poorly mimic the extracellular matrix (ECM) and spatial distribution around the TME, so 3D models are preferred when exploring the intricate crosstalk of cells around the tumour (Fernando et al., 2021). Recently, hydrogel-based 3D TME recapitatilation has proved itself as an excellent resource for in-vitro tumour modelling (Rodrigues et al., 2021). Hydrogels are networks of cross-linked polymers that are “swollen” by water; they are a great in vitro model, as they have the capacity to recapitulate ECM, model protein adhesion and sequesterilization (Caliari et Burdick, 2016). As a part of our Integrated Human Practices efforts, we have interviewed Dr. Anne-Marie Fortier from McGill, who works with hydrogel for precision oncology and kindly consulted us on hydrogel experimental protocol optimization. We have concluded that for our purposes, Polyethylene Glycol (PEG) hydrogel will be optimal. We will use the hydrogel as a biological scaffold to seed with murine TME cells. The cells will be requested from UBC labs that are investigating tumourigenesis in a murine model and are willing to share the TME cell suspensions from hot and cold tumours. The following protocol will be implemented:
1) To add peptide sequences to polymer, react peptide of interest with DA-PEG-NHS in 50mM Sodium Bicarbonate (pH 8.5) for 2hrs. Peptide:PEG = 1:1 molar ratio.
Final concentration of peptide on PEG hydrogel should be between 1.4-5 mM
For 100 uL PEG hydrogel, 3 mM RGDS, 0.13 mg RGDS (65 uL of 2 mg/mL stock), 1.02 mg PEG-NHS
3) Lyophilize mixture (O/N) and store frozen, protected from light.
4) Combine 0.2g/ml PEGDA+PEG (in desired ratio and total weight percent for mechanical compliance) and 0.3mmol/ml triethanolamine in 50mM Tris (pH 8.5).
For 100 uL (final) of PEGDA solution: 20 mg PEGDA+PEG, 2.8 mg triethanolamine-HCl (F.W. 185.65), 100 uL 50mM Tris
5) Add acryloyl-PEG-RGDS and photoinitiator (4 uL into 100 uL polymer of 2,2-dimethyl-2-phenyl-acetophenone (DPA) in n-vinylpyrrolidone (VP) at 600 mg/mL).
6) Sterilize entire polymer solution through 0.2um filter into appropriate tissue culture dish.
7) Add cell suspension to this polymer solution to create cell-polymer solution with a cell density of 3-5 x 106 cells/ml.
8) Photopolymerize with UV light (1-10 min).
9) At this point, 3-D constructs can be transferred to larger well-dish and surrounded with growth media to incubate.
Upon the seeding of the hydrogel, we plan to co-culture it with the Salmonella transfected with pSicA-LuxABG and pLactate-LuxCDE as per the Johnston et al. protocol. Salmonella will then be isolated and inspected under the plate reader for luciferase activity.
INDUCIBLE PROMOTER DISCOVERY
To hunt for inducible promoters in the Salmonella genome, we implemented a promoter trap adapted from the design from UBC iGEM 2019 and Bumann and Valdivia (2007). In a promoterless vector, we inserted a restriction site (BamHI) upstream of a forward reporter (GFP) and a reverse reporter (RFP). The restriction is where fragments from the sheared Salmonella genome will be inserted. If this inducible promoter happens to have directionality and regulates in the forward direction, it will drive the expression of GFP, and if it regulates the reverse direction, it will drive the expression of the reverse RFP. These results can be sorted by colour, resulting in candidate inducible promoters that can be further validated through sequencing.
Even though we did not reach the assay development stage of this section of our project, we have outlined how we would carry these assays out. To produce the sheared Salmonella genome, we will follow the protocol outlined in Bumann and Valdivia (2007), involving the following procedures:
1) Genomic isolation from untransformed Salmonella SL1344 ∆invA∆ssaR.
2) Gel electrophoresis of genomic isolation products to confirm that genomic DNA is not smeared or cleaved.
3) Sonication of bacteria in 10 second intervals for 30 seconds, then transferring 4ul aliquots of DNA from the sonication to a 1.5% gel to observe size of fragments. Sonication will be repeated until gel shows fragments of 500-700bp. This length is large enough to generously capture promoter and regulatory elements of the genome.
4) Gel purification of gel segments from the sonication that resulted in 500-700bp fragment.

Once our genomic fragments have been produced, 20ul ligations to our previously BamHI-digested construct will ensue, leaving at 4C overnight. To screen for a regulatory element, we will produce a stable line of Salmonella strain SL1344 ∆invA∆ssaR transformed with the screening construct, whose colonies will be kept as glycerol stocks in -80C aliquots for future use. From a streaked iteration of this line, we will discard any fluorescent colonies through cell sorting since they would be demonstrating non-specific fluorescence that fails to be regulated by our screening construct.
Since induction of psicA by TNFa has been reported (Ma et al, 2010) but occurs through an unknown mechanism, hence decreasing the reliability of this correlation to be used by other researchers, we decided to screen for other TNFa-inducible promoters in Salmonella genome. Additionally, obtaining psicA in our screening results from this assay would serve as a proof-of-concept that our assumption about the relationship between TNFa and psicA is correct. For this same reason, we chose to test for induction with lactate as a proof-of-concept as well. Since hypoxia is a key characteristic of the tumour microenvironment and a feature that naturally localizes Salmonella to tumours in the first place (Chien et al, 2017), we decided to also screen for promoters that are inducible by hypoxia through incubating Salmonella with our screening vector in an anaerobic chamber previous to cell sorting. Even though we would initially screen for inducible promoters using these two conditions, we would expand to other conditions once these were completed as proof-of-concept.
Cell sorting would be carried out using a Salmonella strain SL1344 ∆invA∆ssaR transformed with an empty vector as a negative control, and transformed with a constitutively expressed RFP as a positive control. Sorting would be based on fluorescence levels of both green and red from reverse RFP and forward GFP expression, respectively. Once sorted, cells with highest fluorescence would be grown overnight with the same volume of its induction molecule (or in the hypoxic chamber), and sorted in the same manner the next day to ensure induction was not a false positive. Cells that have been selected on both rounds of sorting will be plated, and plasmid DNA purified to further characterize candidate gene fragments that induced fluorescence.
[1] Fernando, K., Kwang, L. G., Lim, J. T. C., & Fong, E. L. S. (2021, April 7). Hydrogels to engineer tumor microenvironments: In vitro. Biomaterials Science. Royal Society of Chemistry. https://doi.org/10.1039/d0bm01943g
[2] Rodrigues, J., Heinrich, M. A., Teixeira, L. M., & Prakash, J. (2021, March 1). 3D In Vitro Model (R)evolution: Unveiling Tumor–Stroma Interactions. Trends in Cancer. Cell Press. https://doi.org/10.1016/j.trecan.2020.10.009
[3] Caliari, S. R., & Burdick, J. A. (2016, May 1). A practical guide to hydrogels for cell culture. Nature Methods. Nature Publishing Group. https://doi.org/10.1038/nmeth.3839
[4] Johnston, T. G., Yuan, S. F., Wagner, J. M., Yi, X., Saha, A., Smith, P., … Alper, H. S. (2020). Compartmentalized microbes and co-cultures in hydrogels for on-demand bioproduction and preservation. Nature Communications, 11(1). https://doi.org/10.1038/s41467-020-14371-4