Team:Rio UFRJ Brazil/Results
Results
Overview
With the coding sequences designed (for more details see the Design and Engineering success pages), we requested the synthesis of genes cloned in the pET-28(a) vector by Genscript. With the plasmids in hand, we need to define a workflow for the production, purification and testing of this protein.
Production
For production, it was necessary to transform the inserts in our chassis, the E. coli Bl21(DE3), to carry out the cultivation, induction and expression of the proteins. Unfortunately, due to the pandemic and restrictions on access to the laboratory during late 2020 and early 2021, we were unable to perform some of these experiments and chose to hire the service of Protein Advanced Biochemistry (PAB), a laboratory from the UFRJ specialized in recombinant protein expression, execution the transformation of coding inserts for DME-C and DME-BR in E. coli.
PAB was also responsible for the expression and purification of DME-BR, so we will not include these experiments here and we will focus on the steps of cultivation, induction, expression and purification of DME-C.
Purification
As described on the design page, we have chosen to include a 6xHis-tag at the carboxy-terminal end of both DME-C and DME-BR to facilitate their purification process via immobilized metal affinity chromatography-IMAC. As best described in the sessions, it took additional purification steps to solve some issues we had.
Immunoassays
The main objective of our project is to identify anti-Dengue antibodies in a specific way, preventing or minimizing the recognition of anti-Zika antibodies. For this, we first need to verify through western blot assays whether our protein was in fact capable of recognizing anti-Dengue antibodies. Subsequently carry out anti-Zika antibody recognition assays to check for cross-reactions. And finally, mimic ELISA assays with commercial antibodies to partially assess the sensitivity of the protein we designed.
Production of multi-epitope proteins
Plasmid confirmation
Once we received the plasmids, with the help of the PAB, we cloned them in E.coli BL21(DE3) and performed a miniprep for plasmid extraction.
Expression and purification of DME-BR
As previously mentioned, PAB was responsible for the expression and purification of the DME-BR protein. A titer of 31.3 mg/L of DME-BR was obtained, with an estimated purity of 97.5% ± 1.4. During expression, they reported that under non-denaturing lysis conditions, the protein aggregates and therefore to solubilize it it was necessary to add dithiothreitol (DTT) during the lysis and purification steps. At the end, the protein was stored in a solution of 50 mM sodium phosphate, 250 nM NaCl, 5 mM DTT, pH 8.0.
Expression test and DME-C solubility test
For the expression of DME-C, E. coli BL21 (DE3) was transformed with the constructs by heat shock method, with the aid of PAB. The next steps of culture growth and protein purification were carried out by our team. Transformed positive clones were grown in liquid LB medium with kanamycin (30 μg/mL) as a selection marker, and protein expression was induced with 1mM IPTG during 4 hours. The culture conditions were adapted from the work of Anandarao and collaborators (2005 and 2006), who also expressed multi-epitope recombinant proteins in E. coli [1,2]. Aliquots of culture (1mL) were collected before and after induction, after, 2h and 4h. Cells were centrifuged, resuspended in native (50 mM sodium phosphate and 300 mM NaCl) or denaturant (50 mM sodium phosphate, 300 mM NaCl and 8M Urea) lysis buffer, and lysed by sonication. We tested these two lysis conditions to assess the need for a denaturing agent to keep the proteins soluble.
Analysis of the protein profile by 15% SDS-PAGE showed a band below 26 kDa, possibly corresponding to DME-C (16.83 kDa) in samples collected after 2 and 4 h of induction, both in the soluble fraction and in the insoluble fraction, when using buffer without urea. When the denaturing agent urea was added, the band was observed only in the soluble fraction (Figure 2). Since part of the protein was found in the soluble fraction without urea, we chose to follow the next lysis experiments in native lysis buffer without urea.

Small-Scale DME-C Purification
To confirm the presence of DME-C, we first performed a small scale purification using the His Microspin Purification kit (Amershan Biosciences) according to the manufacturer's instructions and analyzed the different fractions by both 15% SDS-PAGE and Western Blot anti-his. Analysis by 15% SDS-PAGE showed the presence of some contaminating proteins of higher molecular weight in the eluate, which
highlighted the need for further additional purification steps (Figure 2).

Western blot anti-his confirmation from DME-C and DME-BR.
Regarding the western blot, different dilutions of uninduced samples, induced by 0h and 4h (lysis without urea) and of the eluates obtained in the purification were used in order to avoid saturation of the detection signal. Anti-mouse monoclonal antibody 6x-His-Tag (Invitrogen) at 1:2000 dilution was used, and as a secondary anti-mouse antibody conjugated to HRP, at 1:10,000 dilution. The analysis revealed the presence of DME-C, since the anti-His-Tag antibody was able to recognize the over-expressed band with a molecular weight close to that expected. Furthermore, it was observed that one of the bands with the highest contaminating molecular weight present in the purification eluates was also recognized in the assay. As this band was not present either in the uninduced sample or in the sample with 0h of induction, it possibly corresponds to a protein aggregate of DME-C (Figure 3).

Regarding DME-BR, a western blot analysis was performed using anti-6 HIS-TAG antibody (1:2000 dilution) and developed with anti-mouse antibody conjugated to HRP as the secondary antibody (1:10,000 dilution) using ECL (Amersham)
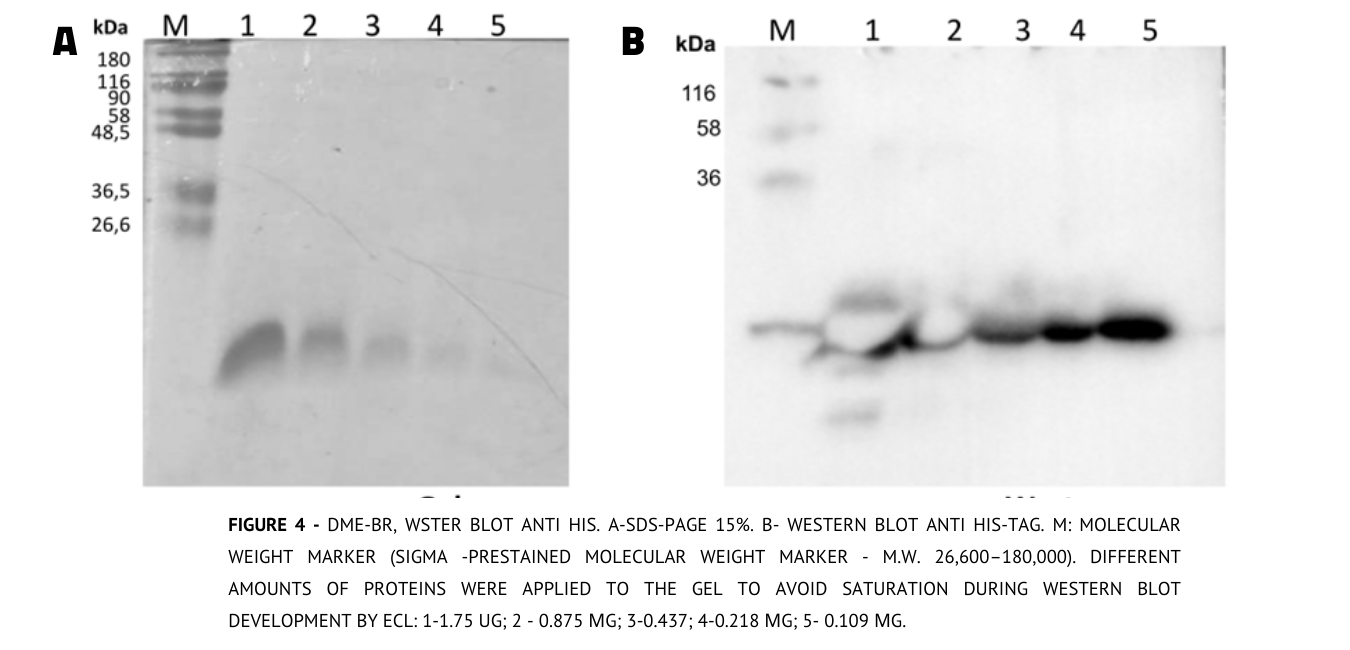
Regarding DME-BR, a western blot analysis was performed using anti-6 HIS-TAG antibody (1:2000 dilution) and developed with anti-mouse antibody conjugated to HRP as the secondary antibody (1:10,000 dilution) using ECL (Amersham)
Expression in larger volume
Cultivation and induction
Once the expression of DME-C was confirmed by the western blot, we expressed the protein in a larger scale (500 mL of LB medium and 30 ug/mL kanamycin), following the same conditions used for small-scale expression. To improve the lysis yield we tested the addition of Cellytic (Sigma Co), to the previously used lysis buffer (without the addition of urea) to resuspend the cell pellet equivalent to 40 ml of culture. By analyzing the 15% SDS-PAGE gel of these lysates, we verified that the protein yield was higher in the soluble fraction using Cellytic, so we chose to continue the use of this reagent.

Purification
After lysis, the supernatant was filtered using a 45 μm filter and we proceed to perform the affinity chromatography using a His-Trap FF. The whole protocol and method used can be checked on the Experiments page. We tested 3 different elution methods, in order to evaluate changes in the chromatographic profile.
CV - Colum Volume
At the last run, we decreased the final elution concentration because we had imidazole interference at the used absorbance wavelength. In the end, not many differences were observed among the three performed analyses and fractions enriched with DME-C proteins were obtained (Figure 6).

| Parameters | First run | Second run | Third run |
|---|---|---|---|
| Equilibration volume | 2 CV | 3 CV | 5CV |
| Sample | 30.0 mL | 35.0 mL | 35.0 mL | Buffer B gradient | 0 - 100 % | 0 - 100 % | 0 - 60 % |
| Elution volume | 6 CV | 8 CV | 6 CV |
At the last run, we decreased the final elution concentration because we had imidazole interference at the used absorbance wavelength. In the end, not many differences were observed among the three performed analyses and fractions enriched with DME-C proteins were obtained (Figure 6).
Diafiltration
Diafiltration is a technique that can remove or lower the concentration of salts like imidazole from the medium by the use of ultrafiltration membranes. We used a 5Kda membrane to retain the DMEC protein and washout imidazole. We pooled the following fractions from the three different elutions from His-trap columns where we obtained the greatest visibility of the bands, as previously shown in Figures 6:
1st (F4-F7), 2nd (F3-F6), and 3rd run (F3-F6),.
After diafiltration we measured the protein concentration by absorbance at 280nm using the Diodrop instrument. The parameters used were: molecular weight (16.83 kDa) and extinction coefficient (1948 M-1 cm-1), both calculated at https://web.expasy.org/.
The absorbance of concentrated protein in diafiltration at 280nm was 0.596 which is equivalent to 0.202μg/μl.
The absorbance of concentrated protein in diafiltration at 280nm was 0.596 which is equivalent to 0.202μg/μl.
Immunoassays
Anti-dengue assays
In order to evaluate if our proteins could recognize anti-Dengue antibodies, we performed western blot analysis with two different commercial antibodies: anti-Dengue NS1, and anti-Dengue envelope protein. Fortunately, both DME-C and DME-BR were capable of recognizing anti-Dengue E antibodies, showing that Ammit fullfiled at least its main function. However, we still needed to evaluate its cross reactivity.

Anti-Zika assays
Considering that our project is aimed at cross-reaction mitigation, we performed a western blot with Zika virus NS1 antibodies, so we could measure if our multi-epitope proteins designed for Dengue infection diagnostics were recognizing anti-Zika antibodies. When analyzing the membrane with ZiKa antibodies, it is possible to observe a band, which should not occur, since the protein was developed in order to avoid cross-reaction.
Western blot anti-Zika NS1 confirmation from DME-C and DME-BR
Regarding this Western Blot, different dilutions of DME-C and DME-BR protein were used. For this purpose, the Anti Mouse monoclonal antibody was used at a 1:10,000 dilution with the Anti-Zika NS1 at a 1:1000 dilution. The analysis revealed the presence of DME-C and DME-BR, since the anti-Zika NS1 antibody was able to recognize the very apparent band with the approximate molecular weight of the anti-histag. As the experiment was performed on the same membrane that was used to perform the anti-histag Western Blot, a wash-related error may have occurred, at the time of stripping the antibodies were not completely removed and therefore when the Zika membrane was revealed the result was not what was expected.

Final conclusions
Observing the analyses, we concluded that to minimize the detected cross-reaction, as shown in the anti-Zika NS1 Western blot, we should iterate more times over the build test and learn design cycle. However, a very valid option would also be to test with antibodies against other proteins.
References
1. AnandaRao R, Swaminathan S, Fernando S, Jana AM, Khanna N. A custom-designed recombinant multiepitope protein as a dengue diagnostic reagent. Protein Expression and Purification. 2005;41(1):136–47.
2. AnandaRao R, Swaminathan S, Fernando S, Jana AM, Khanna N. Recombinant multiepitope protein for early detection of dengue infections. Clinical and Vaccine Immunology. 2006;13(1):59–67.
3. Molina F, Rueda A, Bosque-Sendra JM, Megias L. Determination of proteins in the presence of imidazole buffers. Journal of Pharmaceutical and Biomedical Analysis. 1996;14(3):273–80.
4. Qiagen. FAQ [Internet]. QIAGEN. [cited 2021Oct17]. Available from: https://www.qiagen.com/no/resources/faq?id=6a331f4d-8716-4adf-992c-298fdddbb0be&lang=en
2. AnandaRao R, Swaminathan S, Fernando S, Jana AM, Khanna N. Recombinant multiepitope protein for early detection of dengue infections. Clinical and Vaccine Immunology. 2006;13(1):59–67.
3. Molina F, Rueda A, Bosque-Sendra JM, Megias L. Determination of proteins in the presence of imidazole buffers. Journal of Pharmaceutical and Biomedical Analysis. 1996;14(3):273–80.
4. Qiagen. FAQ [Internet]. QIAGEN. [cited 2021Oct17]. Available from: https://www.qiagen.com/no/resources/faq?id=6a331f4d-8716-4adf-992c-298fdddbb0be&lang=en
© Osiris