In order to build Ammit, we used the Design, Build, Test, and Learn (DBTL) cycle for both the selection of epitopes and their combination. The idea is to use a rational and systematic approach to develop more specific antigens for serodiagnostics. Although the DBTL cycle is commonly used for biological circuits design and metabolic engineering, here we show its potential application for protein engineering. As a proof of concept, we decided to test our methodology for designing a multi-epitope protein for the detection of antibodies present during Dengue 2 (DENV2) infections, since this is the main circulating dengue serotype in our country Brazil. So we can separate our strategy in two different DBTL cycles:
Epitope Selection
Protein Design
When choosing epitopes for serodiagnostics, some considerations are needed:
Unlike multi-epitope proteins used for vaccine development, which uses both B-cell epitopes and T-cell epitopes in its constitution, serodiagnostic multi-epitope protein uses only B-cell epitopes.
Those epitopes must be immunodominant;
Usually, linear epitopes are preferred over conformational ones.
When a chimeric protein is designed, mostly we cannot guarantee that each domain structure will remain the same as it is in the native protein. Besides, linear epitopes are easier to predict and screen than conformational ones. Therefore, the first step of our project was to select which linear B-cell epitopes to use for recognizing anti-dengue antibodies in a specific manner. After epitope selection, the next challenge was to assemble them and check the final protein physico-chemical properties and surface exposure for interacting with antibodies. This way, each iteration throughout the Epitope selection and Protein design cycles should theoretically provide more specific antigens for serodiagnostics.
DBTL Cycle 1 - Epitope selection
First iteration:
Design & Build - Literature review
The studies regarding the use of Multi-Epitope proteins for serologic diagnostics usually screen experimentally synthetic peptide libraries using infected patient samples in order to evaluate the most promising epitopes in terms of sensibility and specificity [1]. Another option, especially when the pathogen is an emerging one such as the SARS-CoV-2 is to predict B-cell epitopes using bioinformatic tools such as Discotope.
Since there are already characterized Dengue epitopes, we have decided to start with known epitopes described in previous studies.
In an initial literature research, we found a study that had already designed a multi-epitope protein for capturing anti-Dengue virus (DENV) IgG by joining 15 linear B-cell epitopes from different DENV proteins and serotypes, as shown in the table below [2,3]. They have chosen epitopes from Envelope protein, since it is the most immunogenic of all DENV proteins and has well characterized immunodominant epitopes. Also, epitopes from the Non structural proteins 1 and 3 (NS1 and NS3 respectively) were used since they can elicit strong antibody responses, especially during secondary infections [2,3]. It is important to note that secondary infections (that is infection of individuals seropositive for DENV due to prior exposure) are really important to be aware of since they are usually associated with complications such as dengue hemorrhagic fever and dengue shock syndrome.
However, the authors have used sequences from Asian strains and did not evaluate the possibility of cross-reaction against other flaviviruses such as the Zika virus (ZIKV). Still, since they have used this protein for the development of a lateral flow assay that presented good results, we decided to start our initial epitope pools with the ones they used previously.
Table 1 - Epitopes used by Anandarao [2,3]
Epitope
Protein
Sequence
Ep1
Envelope
ETLVTFKNPHAKKQDVVVLGS
Ep2
Envelope
NLLFTG
Ep3
Envelope
PFGDSYIIIGVE
Ep4
Envelope
QLKLNWFKKGSS
Ep5
Envelope
TAWDFGSLGGVFTSIG
Ep6
Envelope
VIITWIG
Ep7
Envelope
STSLSV
Ep8
Envelope
VTLYLGA
Ep9
NS1
EHKYSWKS
Ep10
NS1
DSGCVVSWKNKELKC
Ep11
NS1
KFQPESPARLASAILNA
Ep12
NS1
LKYSWKTWGKAK
Ep13
NS1
FLIDGPDTSECPNERRA
Ep14
NS1
WYGMEIRPLSEKEENMV
Ep15
NS3
ILEENMEVEIWTREGEKKKL
Test - Evaluating criteria for cross-reaction minimization
Linear alignments
The first criteria we used to evaluate the epitopes was conservation among the DENV Brazilian strains. We compared the epitope sequences to 13 Brazilian DENV 2 strains that circulated in the country between 2008 and 2019 (GenBank: AGK36289.1; AGK36290.1; AGK36293.1; AGK36296.1; AGK36295.1; AGK36299.1; QGW05368.1; QGW05372.1; AGK36292.1; AGN94882.1; QGJ02362.1; ACY70783.1; QGW05381.1). Using the multiple alignment software tool from Clustal server (https://www.ebi.ac.uk/Tools/msa/clustalo/), we identified some differential residues in the epitope sequences that were conserved within Brazilian strain sequences. These residues are highlighted in blue in the sequence below.
Figure 1 - Differential residues between AnanadaRao sequence and Brazilian Strains.
Also, you can download the alignment file in the button below.
As the aim of the present work was to express a multi-epitope protein with greater specificity for recognizing antibodies during infection by Brazilian DENV2 strains, epitopes 2, 4, 5, 6, 7, 8 and 12 from table 1 were not used for the construction of Ammit because they do not present differential residues. Epitope 1, despite not having differential residues either, was kept as it was pointed out as a potential immunodominant diagnostic peptide specific for Dengue in a previous bioinformatics study [5].
We decided to evaluate if changing those differential residues would cause any impact on antibody recognition. Therefore, we decided to build two multi-epitope Ammit proteins: DME-C, which will use the epitopes we select during the first DBTL cycle; and DME-BR, which will be constituted by the same epitopes, but with some few mutations in order to make the sequence closer to Brazilian strains.
After analysing the differential residues, we decided to change only those in which the mutation would be for another residue with similar polarity. Changes between a neutral polar residue and a charged one were not made. Check below the changes:
Conservancy analysis
To verify epitopes that could cross-react with other arboviruses, we analysed the epitopes selected in the previous step in the Epitope Conservancy Analysis tool, available in the Immune Epitope Database (http://tools.iedb.org/conservancy /) (BUI et al., 2007). For this analysis, the epitopes were compared with sequences from 41 yellow fever (YF) and Zika virus strains circulating in Brazil between 2008 and 2019.
Check in the buttons below to check the Zika and YF strains used.
The software generates a table that provides information about the maximum and minimum identity found among the alignments and also the percentage of sequences that had identity higher than a preset identity threshold [6].
According to some studies, 60% [7] to 80% [8] of identity can be found in highly conserved flavivirus regions, which might correlate with cross-reactivity among this virus family. In order to be more restrictive, we decided to use the 60% as a threshold.
Table 2 - Conservancy analysis with Zika and Yellow fever sequences
Epitope
Percent of protein sequence matches at identity <= 60%
Minimum identity
Maximum identity
Ep1
78.05
42.86
71.43
Ep3
100
75
83.33
Ep9
0.00
37.5
50
Ep10
0.00
46.67
46.67
Ep11
0.00
41.18
47.06
Ep13
0.00
47.06
52.94
Ep14
78.05
52.94
76.47
Ep15
0.00
35.00
45.00
Structural alignments
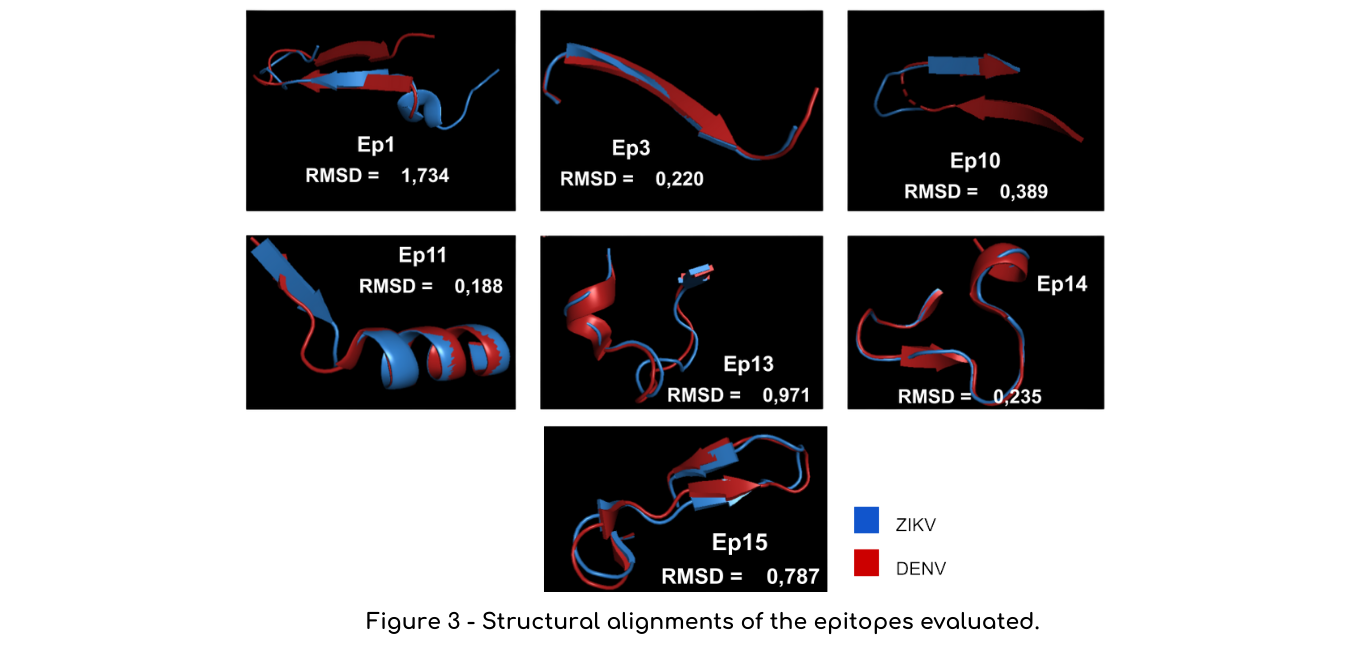
Another analysis we performed were structural alignments in the software Pymol between the epitope structure in the Dengue native protein (PDB code: Envelope, 1OKE; NS1, 4O6B; NS3,2VBC) e and their corresponding structure in Zika virus (PDB code: Envelope, 5JHM; NS1, 5GS6, NS3, 6ADW). Unfortunately, epitope 9 did not have its structure elucidated, so we could not run this analysis for this sequence. We measured the Root-mean-square deviation of atomic positions (RMSD) to evaluate the structural similarities between those regions. The closer the RMSD is to zero, the more similar are the structures (Figure 3).
Researchers feedback
Besides the analysis we have talked to researchers in order to improve our design choices:
We confirmed the importance of using B-cell epitopes for serodiagnostics after talking to the researcher Adriana Vallochi, from Fiocruz, who has expertise with immunology. As mentioned in the project description, the B-cells are the ones associated with the humoral immune response, producing antibodies when stimulated by an antigenic region: a b-cell epitope. Those epitopes are also recognized by antibodies, and that is their importance for serodiagnostic [4].
Also, we talked to Professor Marcius Almeida, who recommended us to also evaluate if the epitopes we analysed had cysteine residues forming disulfide bonds. According to him, those bonds might help stabilize the protein structures, which might impact their proper folding in chimeric proteins, so we should be careful. After analyzing the epitope structures in Pymol, we have identified that epitopes 10 and 13 had disulfide bonds in their structure.
Learn - Discarding problematic epitopes
Based on those analyses, we decided to eliminate the epitopes that both presented bad conservancy results and a low RMSD: epitopes 3 and 14. According to literature, RMSD values below 2.0 Angstrom already indicate that the proteins have very similar structure, so we thought combining both sequence and structural identity instead of only using RMDS values or identity values would be a more robust approach to filter epitopes. Regarding the epitopes containing disulfide bonds, we decided to keep them only because we are using linear epitopes which can be recognized even when denatured. However, knowing which epitopes have this bond might be important in case problems are identified during the final protein expression and folding.
Second iteration:
Design & Build - Finding a new epitope
After another round of literature research, we found a study that identified potential epitopes to use for differentiating dengue from zika. After using a cohort of human sera, the authors highlighted the potential of the envelope protein epitope YENLKYTVIITVHTGDQH, mentioned as peptide 17 in the study, for detecting Dengue specific antibodies [1]. After a multiple alignment with the 13 DENV2 strains used in previous alignments, some mutations were made in this peptide so that its sequence would be close to the sequence present and conserved in Brazilian strains, obtaining the peptide PENLEYTIVITPHSGEEH.
Test - Linear and structural alignments
The conservancy analysis comparing with the previous Zika and Yellow Fever of this epitope showed that it had a maximum identity of 44%, highlighting its potential.
Table 3 - Conservancy analysis for peptide 17
Epitope
Percent of protein sequence matches at identity <= 60%
Minimum identity
Maximum identity
Ep1
0.00
38.89
44.44
Regarding structural alignments, it showed an RMSD value of 1.744, higher than all the other epitopes tested.
Learn - Incorporating a new epitope
Considering the promising linear and structural alignments, we decided to incorporate this epitope in our design.
DBTL Cycle 2 - Protein Design
First iteration:
Design - Epitope and linker assembly
Once we chose the epitopes, we had to decide the order of epitopes and the linkers we would use to join them. Initially, we considered the linkers below, commonly used for vaccine multi-epitope design.
AAY
GPGPG
GGG
Linker selection is important, due to its role in ensuring protein stability and facilitating epitope presentation [9]. Although glycine linkers are commonly used flexible linkers, less flexible linkers such as AAY and GPGPG have their relevance, since they can help preventing the formation of junctional epitopes, created by the juxtaposition of epitopes, which might cause the redirection of the immune response (and also antibody recognition) to undesired regions in the sequence [9]. For deciding which linker to choose, we again consulted Professor Marcius Almdeia, who advised us to use the proline linker, since they can also help stabilize the protein structure.
Regarding the epitopes order in the assembly, we decided to keep the original order used by Anandarao and collaborators, keeping only the selected epitopes and replacing the third epitope by peptide 17. At the end, we designed two Ammit multi-epitope proteins: DME-C, with the chosen epitopes with their unchanged sequences, and DME-BR, which contains some mutations highlighted in order to make the sequence more identical to the conserved Dengue Brazilian strain proteome.
Build - Structure model
In order to evaluate the protein design, we used the PSIPRED server DMPfold tool to predict the 3D structure of the proteins (Check the modeling page for more details).
Test - Structure validation and antibody simulations
Using the protein primary structure, we build hydrophobicity plots in order to evaluate the presence of exposed regions in the protein, which would thus be able to interact with the antibodies. Fortunately both DME-C and DME-BR presented exposed regions.
Regarding the 3D structure validation, we have generated Ramachandran plots that indicated the presence of a high percentage of residues in preferred regions, which shows the quality of our prediction.
Finally, we used the 3D structures we have generated in order to make antigen-docking simulations.
We were not considering performing docking since we only used linear B-cell epitopes. However, after the Queens team suggestions, we thought it would be a good idea to check if our protein could bind to anti-Zika antibodies. So we used the software ClusPro and identified epitopes that might be able to cross react with anti-Zika antibodies (Check modeling for more details).
Design - Epitope and linker assembly
Although both DME-C and DME-BR presented exposed surface regions, there is a chance of cross-reaction as the docking simulations presented. However, when we talked to the Queens team who suggested us to use ClusPro, we had already synthesized the plasmid containing the coding sequence for DME-C and DME-BR and unfortunately we could not implement modifications to resolve this matter.
Nevertheless, since our final experiments showed the occurrence of cross reaction, now we can have an idea of the potential epitopes that might be associated with this event and new iterations of both DBTL cycles might improve the specificity of the antigens.
Wet lab implementation
Once we optimized Ammit design for minimization of cross-reaction using the DBTL cycle, we defined cloning strategy (Check Design for more information), and proceeded to the experiments needed its expression in Escherichia coli and finally the immunoassays to validate Ammit’s capacity of recognizing antibodies in a more specific way.
Check out Experiments and Results for more detail.
References
1- Amrun SN, Yee WX, Abu Bakar F, Lee B, Kam YW, Lum FM, et al. Novel differential linear b‐cell epitopes to identify Zika and dengue virus infections in patients. Clinical & Translational Immunology. 2019;8(7).
2- AnandaRao R, Swaminathan S, Fernando S, Jana AM, Khanna N. A custom-designed recombinant multiepitope protein as a dengue diagnostic reagent. Protein Expression and Purification. 2005;41(1):136–47.
3- AnandaRao R, Swaminathan S, Fernando S, Jana AM, Khanna N. Recombinant multiepitope protein for early detection of dengue infections. Clinical and Vaccine Immunology. 2006;13(1):59–67.
4- Sela-Culang I, Kunik V, Ofran Y. The structural basis of antibody-antigen recognition. Frontiers in Immunology. 2013;4.
5- Lee AJ, Bhattacharya R, Scheuermann RH, Pickett BE. Identification of diagnostic peptide regions that distinguish zika virus from related mosquito-borne flaviviruses. PLOS ONE. 2017;12(5).
6- Bui H-H, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics. 2007;8(1).
7- Salvador EA, Pires de Souza GA, Cotta Malaquias LC, Wang T, Leomil Coelho LF. Identification of relevant regions on structural and nonstructural proteins of zika virus for vaccine and diagnostic test development: An in silico approach. New Microbes and New Infections. 2019;29:100506.
8- Xu X, Vaughan K, Weiskopf D, Grifoni A, Diamond MS, Sette A, et al. Identifying candidate targets of immune responses in zika virus based on homology to epitopes in other flavivirus species. PLoS Currents. 2016.
9- X. Chen, J.L. Zaro, W-C, Shen, Fusion protein linkers: Property, design and functionality. Adv Drug Deliv Ver, 65 (10) (2013) 1357-1369.