
Nattokinase Production
Construct Plasmid for Nattokinase Expression
First of all, we extracted the genomic DNA from Bacillus subtilis natto HS01(a generous gift from Dr. Hsueh).
 Then, we successfully cloned aprN from gDNA of Bacillus subtilis natto HS01 by PCR. At the same time, we added XbaI and XhoI cutting site in front of aprN and behind aprN , respectively.
Then, we successfully cloned aprN from gDNA of Bacillus subtilis natto HS01 by PCR. At the same time, we added XbaI and XhoI cutting site in front of aprN and behind aprN , respectively.
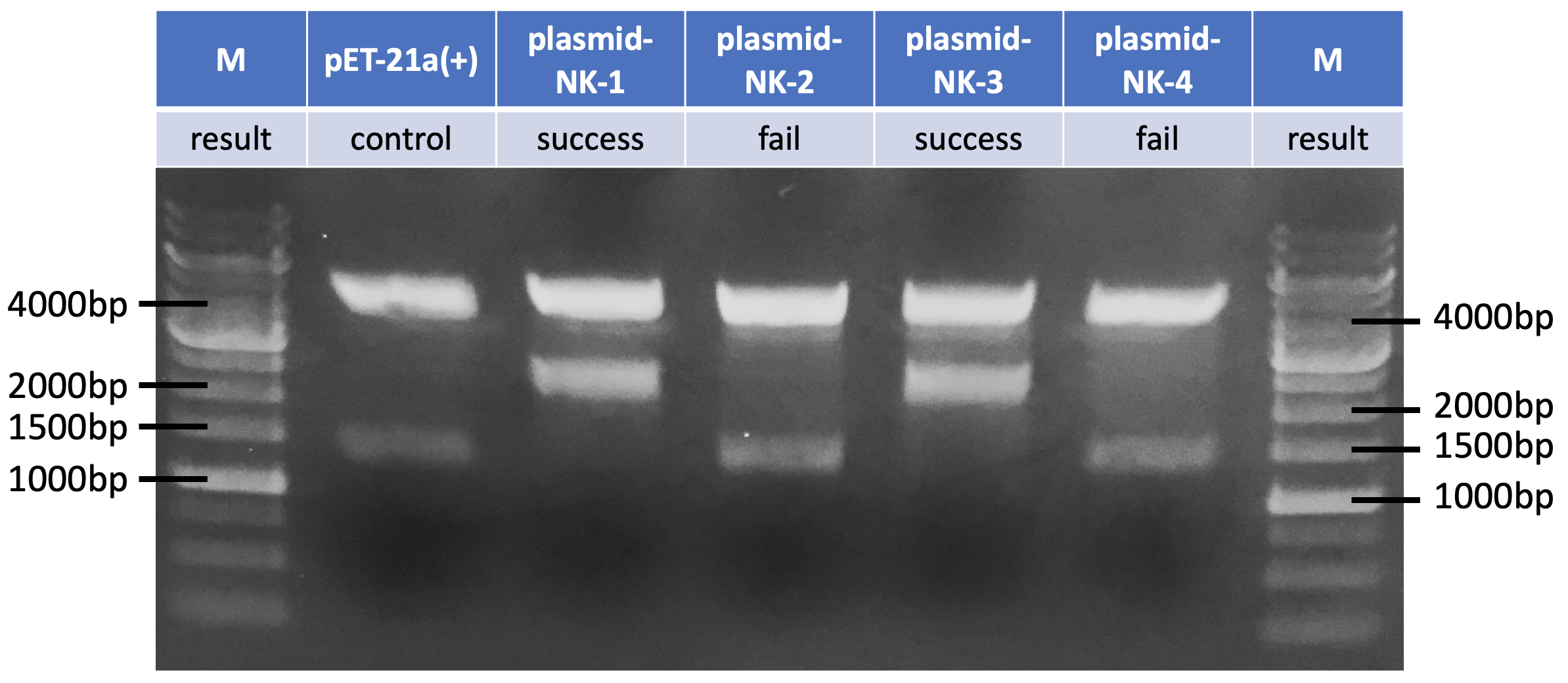
 After ligating aprN PCR product to pET-21a(+), we used NotI/ PstI enzyme digestion to test our construction because there is a NotI cutting site in aprN .
After ligating aprN PCR product to pET-21a(+), we used NotI/ PstI enzyme digestion to test our construction because there is a NotI cutting site in aprN .

 Nattokinase Induction and Test
Nattokinase Induction and Test
To know whether E. coli can produce Nattokinase, we transformed the successfully made plasmid-NK construct into E. coli BL21 cell and treated them with 0.7 mM of IPTG after OD600 of the liquid culture reached 0.6, and measured OD600 every 4 hr throughout the 16 hr induction procedure at 20°C.[1, 2] Next, we checked Nattokinase expression by running SDS-PAGE. We can see there are bands presenting between 25~35 kDa after IPTG induction, and Nattokinase should be around 28 kDa. Thus, there are chances that Nattokinase is successfully expressed.
 However, in our fibrin plate assay, no lytic circle was observed after dropping 10 ul medium onto it, which means no Nattokinase activity was detected. This may be due to the expression level in medium was too low to be detected, or our Nattokinase lost its activity somehow. Also, our SDS-PAGE results of Nattokinase purification were not as expected, too.
However, in our fibrin plate assay, no lytic circle was observed after dropping 10 ul medium onto it, which means no Nattokinase activity was detected. This may be due to the expression level in medium was too low to be detected, or our Nattokinase lost its activity somehow. Also, our SDS-PAGE results of Nattokinase purification were not as expected, too.
 Due to the limitation of our time, we are still in the troubleshooting process. Here are some experiments we plan to do in the near future:
Due to the limitation of our time, we are still in the troubleshooting process. Here are some experiments we plan to do in the near future:
Gene sequencing
Confirm the sequence of aprN sequence and 6xHis-tag are as expected. Make sure our Nattokinase will be produced with 6xHis-tag at the C-terminal.
Western blot
Check the presence of Nattokinase with 6xHis-tag by 6xHis-tag antibody.
Adjust purification protocol and expression condition
In the reference, when E. coli expresses Nattokinase at higher temperature (e.g 37°C, 30°C), Nattokinase will prefer to present at about 40 kDa. On the other hand, while expressing at lower temperature (e.g. 15°C, 20°C), it will present at about 28 kDa (mature protein) instead.[2] So, we are going to check the optimized temperature for our Nattokinase secretion. Also, we may try to use different concentrations of IPTG to induce the expression, add PMSF to avoid unwanted protein degradation (however, this way will permanently inactivate Nattokinase), and so on.
Retest activity
If our Nattokinase is secreted into medium successfully, we can measure its activity by fibrin plate assay and spectrophotometric method to know how our Nattokinase expression level and activity is.
References
First of all, we extracted the genomic DNA from Bacillus subtilis natto HS01(a generous gift from Dr. Hsueh).
Fig 1. The result of Bacillus subtilis genomic DNA extraction.
Fig 2. The result of nattokinase cloning.
Fig 3. Prediction of plasmid-NK digestion(NotI/PstI).
Fig 4. The result of plasmid-NK confirmation.
To know whether E. coli can produce Nattokinase, we transformed the successfully made plasmid-NK construct into E. coli BL21 cell and treated them with 0.7 mM of IPTG after OD600 of the liquid culture reached 0.6, and measured OD600 every 4 hr throughout the 16 hr induction procedure at 20°C.[1, 2] Next, we checked Nattokinase expression by running SDS-PAGE. We can see there are bands presenting between 25~35 kDa after IPTG induction, and Nattokinase should be around 28 kDa. Thus, there are chances that Nattokinase is successfully expressed.
Fig 5. The SDS-PAGE result of Nattokinase induction.
Fig 6. The result of fibrin plate test.
Gene sequencing
Confirm the sequence of aprN sequence and 6xHis-tag are as expected. Make sure our Nattokinase will be produced with 6xHis-tag at the C-terminal.
Western blot
Check the presence of Nattokinase with 6xHis-tag by 6xHis-tag antibody.
Adjust purification protocol and expression condition
In the reference, when E. coli expresses Nattokinase at higher temperature (e.g 37°C, 30°C), Nattokinase will prefer to present at about 40 kDa. On the other hand, while expressing at lower temperature (e.g. 15°C, 20°C), it will present at about 28 kDa (mature protein) instead.[2] So, we are going to check the optimized temperature for our Nattokinase secretion. Also, we may try to use different concentrations of IPTG to induce the expression, add PMSF to avoid unwanted protein degradation (however, this way will permanently inactivate Nattokinase), and so on.
Retest activity
If our Nattokinase is secreted into medium successfully, we can measure its activity by fibrin plate assay and spectrophotometric method to know how our Nattokinase expression level and activity is.
References
[1] Liang X, Jia S, Sun Y, Chen M, Chen X, Zhong J, Huan L. Secretory expression of nattokinase from Bacillus subtilis YF38 in Escherichia coli. Mol Biotechnol. 2007 Nov;37(3):187-94. doi: 10.1007/s12033-007-0060-y. Epub 2007 Jul 17. PMID: 17952663.
[2] Jeong SJ, Cho KM, Lee CK, Kim GM, Shin JH, Kim JS, Kim JH. Overexpression of aprE2, a fibrinolytic enzyme gene from Bacillus subtilis CH3-5, in Escherichia coli and the properties of AprE2. J Microbiol Biotechnol. 2014 Jul;24(7):969-78. doi: 10.4014/jmb.1401.01034. PMID: 24743573.
[2] Jeong SJ, Cho KM, Lee CK, Kim GM, Shin JH, Kim JS, Kim JH. Overexpression of aprE2, a fibrinolytic enzyme gene from Bacillus subtilis CH3-5, in Escherichia coli and the properties of AprE2. J Microbiol Biotechnol. 2014 Jul;24(7):969-78. doi: 10.4014/jmb.1401.01034. PMID: 24743573.






























