Team:MIPT MSU/Results
Introduction
The development of our team is based on three main elements, which were assembled separately in three different plasmids.
The first element is the endogenous retroviral protein syncytin (ERVW-1 gene), which provides targeted fusion of vesicles with cells that have a receptor for this protein (ASCT2).
The second element is the target mRNA with the HIV-1 packaging signal that needs to be delivered to the cancer cell. The packing signal allows the sort mRNA to be packaged into a vesicle. As a target mRNA, we used TurboGFP mRNA (Evrogen - [1]) with b-galactosidase (lacZ) gene, taken from a plasmid kit supplied to us by the iGEM (Plate 2, well: 9E). Thanks to such a sensor, it is possible not only to fix the GFP luminescence, but also to carry out blue-white selection. In other words, this fusion protein allows double control.
Last but not least element of our system is gag-polyprotein, which includes the RNA-binding domain (RBD). This domain recognizes the psi-signal from the second type of constructs, dragging the sensory protein GFP + lacZ along with it.
These three elements, working together, carry out targeted packaging and delivery of vesicles to target cells.
First type of genetic construction - fusion protein syncytin
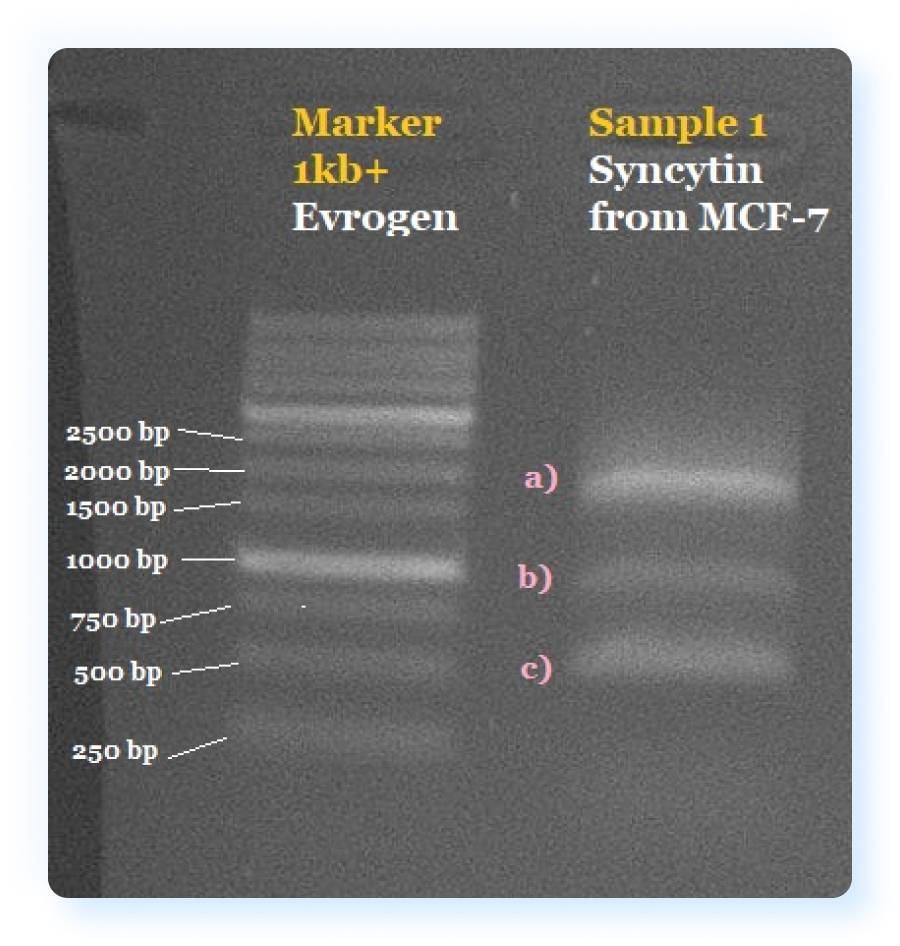
Initially, we tried to obtain syncytin from the MCF-7 cell culture, in which its expression is observed. We isolated total RNA from MCF-7 cells and performed reverse transcription followed by PCR for the ERVW-1 gene. Using electrophoresis, we validated the result of obtaining cDNA of the ERVW-1 gene from MCF-7 cells and three fragments of different lengths were found, one of which resembled the expected length of ERVW-1. We assumed that this is due to the fact that our primers also annealed some other sequences present in the MCF-7 culture, which were also present in the total cDNA. In this regard, it was decided to cut a band corresponding to the length of ERVW-1 from the gel and repeat PCR. However, in the next round of PCR, we also observed three bands corresponding to the same lengths. We believe that this is due to poor separation of these three fragments on the gel, due to which some part of the non-target two genes nevertheless, when excised from the gel, got into the excised piece of gel and was re-amplified. Due to this failure, we decided to order the synthesis of the syncytin gene from Twist Bioscience. Having obtained this gene, we repeated PCR and obtained the required fragment.
As part of the first plasmid, we decided to make two designs. One of the constructs contains a stop codon at the end of the syncytin gene, while the other does not contain this stop codon, and after syncytin comes the Turbo-GFP gene [1] in the syncytin reading frame. Thus, in the first case, we get pure syncytin, and in the second case, a fusion protein consisting of syncytin at the N-terminus and Turbo-GFP at the C-terminus. We cloned these two constructs and confirmed the success of the cloning by electrophoresis.
First type of genetic construction.

The gel electrophoresis results after Syncytin amplification from cDNA obtained from the MCF-7 cell line. a)Top band (weight between 1500 and 2000 bp) potentially present correctly amplified Syncytin (expected weight 1617 bp). b)Presents off target band with molecular weight around 1000 bp. c) Presents off target band with molecular weight around 500 bp. Left column presents a 1kb+ Marker from Evrogen.
The gel electrophoresis results after Syncytin amplification from sequence synthesised by Twist Bioscience. Sample 1 - syncytin amplified by using primers with stop codon. Sample 2 - syncytin amplified by using primers without stop codon. Left column presents a 1kb+ Marker from Evrogen.


Second type of genetic construction - psi-signal with mRNA
The second construction contains a Ψ (psi) signal with the target mRNA (as already mentioned, this is Turbo GFP and b-galactosidase mRNA). Initially, we planned to make one construct here, taking psi-signal from lentiviral plasmids and merging it with the target mRNA. However, after careful consideration we concluded that it is necessary to make 5 structures (read more here), shown in the figure below.
By using them we will be able to show that it is psi-signal that ensures the packaging of the target mRNA into vesicles (however, looking ahead, we did not have time to do these experiments due to the compressed amount of time). We ordered the synthesis of the psi-long gene from Evrogen. All structures of the second type were assembled using Gibson Assembly. We confirmed the success of the assembly of the structures by electrophoresis.
Second type of genetic construction.

The gel electrophoresis results of 5 construction made with Gibson Assembly. Left to right: 1kb+ Evrogen Marker, psi(short)-IRES-GFP-lacZ 1295 bp, psi(short)-GFP-lacZ 747 bp, psi(long)-IRES-GFP-lacZ 1525bp, psi(long)-GFP-lacZ 973bp,IRES-lacZ 1149 bp. All constructions presented by bright white bands.

Third type of genetic construction - gag-polyprotein
The last construcion is a lentiviral plasmid containing gag-polyprotein. This gag-polyprotein, as mentioned at the beginning, includes RBD, to which the psi-signal packing signal is attached. The lentiviral plasmid was chosen as the vector, since we want to carry out a stable transfection of MCF-7. The gag-polyprotein gene was kindly provided by our instructor Evgenii Lunev (Institute of Gene Biology, Russian Academy of Sciences). Successful cloning was also confirmed by agarose gel electrophoresis.
Third type of genetic construction.

The gel electrophoresis results after gag amplification after cloning it into a lentiviral vector. Left column presents 1kb+ Marker from Evrogen. Right column presents an amplified gag (weight 1503 bp).

Due to lack of time for the experimental part (due to the long supply of reagents to Russia, difficulties with syncytin cloning and problems with plasmid extraction kit) we did not have time to do the rest of the planned experiments, but we are not stopping here and will continue working on this project after iGEM-2021. What else we plan to do is described below.
First, following the advice of Marlin company and our Instructor Ivan Sorokin, we want to carry out in-vitro translation and transcription of the second type of our construction. This will allow us to check the overall operation of our system outside the cells and confirm the concept of our work. It will also confirm or deny that the presence of IRES in front of the target mRNA improves protein expression from it.
Second, we plan to transfect the target cells with the plasmids we have obtained. We plan to carry out transfection into MCF-7 cells by electroporation using the Lonze kit. Two days after nucleofection we plan to collect a supernatant containing vesicles and drip it onto HT-29. We expect that after such treatment, GFP and lacZ, which are signaling proteins in our system, will be expressed in HT-29 cells.
Third, we will clean our vesicles with the help of columns and select such protocol parameters so that the vesicle output will be more purified.
Finally, to improve our system, we plan to make a knockdown of the ASCT2 syncytin receptor in MCF-7 cells, which are vesicle producers in our experiment. Thus, we exclude the possibility of reuptake by MCF-7 cells of vesicles, which are budded from themselves.
We also plan to change the GFP+lacZ signaling mRNA to miRNA, with which we will be able to regulate the expression of gene of interest. However, we will be able to start this part only after we confirm that our system works on signaling mRNA. In addition, we plan to replace syncytin with other fusion proteins and thus change the targeting of our vesicles to other cell types.
References
- https://evrogen.com/products/TurboGFP/TurboGFP_Detailed_description.shtml
- https://www.researchgate.net/figure/Caspase-activation-induced-by-different-drugs-on-HT-29-cell-line-was-evaluated-by-the_fig5_232249360
![]()