Team:CSMU Taiwan/Model


Model
Introduction
Modeling has played a critical role throughout the course of our project. Especially with the imposed restrictions on lab access during the pandemic, it has provided a platform for prediction and explanation of our lab theories and experiments.
Our team's modeling can be separated into two parts, namely Circular DNA design and Rolling Circle Amplification (RCA) modeling.
The Circular DNA design comprises of a code that provides users with an optimised sequence of a stable circular DNA with no intramolecular loops upon entry of relevant information, which we will discuss in more detail below.
On the other hand, the rolling circle amplification (RCA) modeling makes use of mathematical models to predict the action of phi29 polymerase, which is a crucial enzyme in the mechanism used in our project, GotCha.
For more information on the two models, please click on the icons below.


Circular DNA Design
Overview
We spend a lot of time and energy on building the model, which plays an important role in our project.
Initially, our team tried to use existing circular DNA sequences in scientific journals while only replacing the Micro-RNA binding site of the circular DNA sequence with the complementary DNA sequences of our target micro-RNAs. However, we were faced with the challenge that there were way too many loops in the predicted DNA structures in this scenario, making the DNA sequence unsuitable for use as the loops may intefere with amplification by polymerase. Despite this, we could not find any softwares out there that generates circular DNA sequences.Therefore, after much consultation with esteemed professors such as Dr. Liu Yu Fan
(Human practices)
, graduate seniors such as Chen Kuan-Lin
(Human practices)
we refered to, we came to a decision to design our own circular DNA sequence a model: Circular DNA Sequence Design, which can be found on Github
here
. Through online DNA secondary prediction websites, we have validated our code and therefore used it to generate our circular DNA probe sequences for capture of both our micro-RNA biomarkers. In order to tailor our circular DNA for our project. For more information on part please refer to
Parts
.We believe that our software can provide a new direction in microRNA detection.
Method
Initially, when we manually designed DNA probes and ran it through online DNA secondary structure prediction websites(RNA Structure[1],UNAfold[2],kinefold[3]), we realised that when there is a segment of three or more complementary base pairs in a single DNA strand, there is a high probability of forming stem-loops. We also realised that with GC bases in particular, two base pairs are enough to form an unstable loop. However, after using DNA secondary structure prediction websites, we discover that the probability of formation of the two complementary bases pairs formed by GC bases is less than 50%. Then we consulted Prof. Yao
(Human practices)
with the results of prediction websites, and she informed us that the above can be ignored in our code design because of low probability.
To ensure GotCha will capture our selected miRNAs as expected and at the same time not detach easily, the Immobilisation Probe and miRNA have to bind accurately to binding sites on the circular DNA probe. Therefore, we designed our Circular DNA sequence such that there are no more than two complementary base pairs with Immobilisation probe and miRNA. This ensures high accuracy in binding of Immobilisation and microRNA to their respective binding sites.
To achieve the above, we created a software with python language named "Circular DNA Sequence Design". Our main principle in this design software is that we used a unit of 3 nucleotides at a time to check if there are complementary bases read in the opposite direction. If there are complementary bases, the sequence of 3 nucleotides will be abandoned. If not, the code will continue to check for the next 3 nucleotides until the total length of circular DNA is reached.
Verify
We used three DNA secondary structure prediction websites RNAStructure[1],UNAfold[2],Kinefold[3] to evaluate the secondary structure and thermodynamic stability of our circular DNA sequences. From the results, we see through less than zero gibbs free energy that the conformation of our DNA sequences are formed spontaneously, and there are no significant loops of more than 3 nucleotides joined by complementary base pairing present. This validates our code.
UNAFold |
|
|---|---|
BBa_K4029010 |
BBa_K4029011 |

|

|
RNA Structure |
|
BBa_K4029010 |
BBa_K4029011 |

|

|
Kinefold |
|
BBa_K4029010 |
BBa_K4029011 |

|

|
Future Work
As the rolling circle amplification mechanism is growing in popularity in the field of synthetic biology and diagnostics, we see the great potential our model can bring to the table in these relevant fields (contribution) . In the future, we aim to continue improving our model towards an even more user-friendly software. We recognise that albeit the cut in time our model brings to the process of circular DNA sequence generation, it still lacks the built-in ability to predict DNA structures and value of free-energy, therefore troubling users to manually take an extra step in checking through another website. Therefore, we hope that in the future, this modeling system can be further built on to increase user convenience and provide an efficient platform for both aspiring students such as fellow iGEMers as well as professionals in the field in DNA sequence synthesis.
RCA Modeling
Overview
Through the planning of our experiments (see Design) , we came across an issue of uncertainty in the rate of RCA reaction. (see Design for RCA mechanism) . Through consultations with professors and graduate students (see Human Practices) , we have decided to derive the rate through establishing mathematical models regarding phi29 polymerase action in the RCA mechanism. Additionally, by using the rate of reaction to estimate fluorescence inte, we fully model the RCA mechanism used in our project. With the above established, this model will complement our experiment and help to establish an optimal condition for RCA efficiency.
Phi29 Polymerase
phi29 DNA polymerase is commonly used in RCA reaction. Its high-speed replication and exonuclease activity facilitates the RCA reaction in running correctly and efficiently[8]. In addition, according to Javier Saturno’s study[8], phi29 polymerase does not dissociate from DNA template. In this case, we concluded that complex dissociation will not influence our experiment data. However, there is no existing literature on phi29 polymerase kinetics in the context of RCA reaction. To model our experiment, we decided to design a model that based on the mechanism of phi29 polymerase.
Polymerase Activity
We used Michaelis-Menten kinetics[4] to model the mechanism of phi29 polymerase. For phi29 polymerase to carry out DNA elongation, it needs to be attached to a DNA strand, and add dNTPs to the 3’ end of the growing strand as a substrate.
Through the characteristics of phi29 polymerase being that :
1. elongation will only happen when miRNA primer is present
2. the DNA template will not dissociate
3. phi29 will reach its best efficiency once activated.
From the above description, we have derived the below equation.

D0 stands for miRNA primer, and Di stands for primer with i-mer dNMP; E•Di stands for the binary complex formed by the i-mer primer DNA and DNA polymerase; N stands for dNTP concentration.
According to the reaction of[8], we assume the miRNA and phi29 polymerase binary complex as an enzyme complex as an enzyme complex [E•D] to represent the activated and reactive enzyme, and [E•D] is controlled by mirna
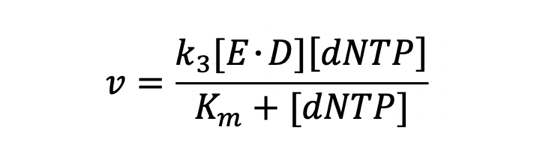
According to the DNA polymerase model of [5], Michaelis-Menten model can used to explain the activity of phi29 polymerase. We chose the data in this scientific paper[7] which is similar to our experimental environment as the reference of k3 and Km(k3=120,Km=31uM), and we adjusted the Michaelis-Menten kinetics equation according to our needs. We have set the dNTP concentration to be 2mM
(experiment)
ensure that the concentration of dNTP be excessive. This way, the phi29 polymerase replication activity is promised.
According to the above formula and parameters, we use matlab predicted phi29 polymerase reaction rate

DNA Concentration

Dn stands for RCA product with n-mer nucleotide
According to the equation above we derived a speed value (v), and through the product of speed (v) and time (t) we derived the amount of total DNA concentration in fixed time (Dn).
Result
Using our mathematical formula established, our team has managed to formulate the relationship between fluorescence absorbance and RCA reaction. We designed three variables, namely: E.D, dNTP, t, in order to more accurately model the action of phi29 in our mechanism, and from there derive the product DNA concentration.

Conclusion
After comparing our model with our experiment results, we realised that there are certainly disparities between the two. Through further research, we have realised that we failed to take into account factors such as metal ions and exonculease activity in phi29 polymerase. However, due to time constraint, we were unable to conduct further verifications in the above factors and formulate more relationships to justify the differences.
According to Robert J. Bauer et al.[6] Metal ions are also a major factor affecting the action of polymerase.Km,K3 is the Michaelis constant, and [M2+](Mg2+ or Mn2+) is the concentration of the divalent cation included in the reaction. Data obtained from M2+ titration reactions were fit to a Michaelis-Menten equation with a cooperativity (n) parameter:

For those reactions where inhibition was observed at higher concentrations of M2+, an inhibition reaction equation was used instead:

Ki is the inhibition constant.
According to Cristina Garmendia's research[8], exonuclease activity of phi29 polymerase will certainly affect the reaction rate of phi29 polymerase.

They derived the rate formula affected by exonuclease activity through the equation above.


In these formulae, Di and (Di•N) represent the concentration of a certain DNA intermediate, i-bases long, and the complex i-mer DNA primer with the next nucleotide to be incorporated bound in its position, not yet catalyzed (see Fig. SI); both Di and (Di•N) stand for the corresponding complexes with DNA polymerase; Dt is the total amount of DNA polymerase/DNA complexes; N stands for dNTP concentration, and Km = (k-1+ kcat)/k1.For more details, please see Cristina Garmendia's research[8]
Apart from the above mentioned, we are aware that there are still variables such as temperature and other factors that influence enzyme activity that we have yet to take into account. However, the above factors mentioned still lack extensive in the context of RCA, and therefore we have decided to forgo them for the competition due to time constraint and constraints by the covid-19 pandemic. We hope that after the pandemic eases, we will be able to conduct more experiments to account for the gaps between our model and experiment results.
References
- Iman Jeddi, Leonor Saiz.Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors.Sci Rep 2017;7(1):1178.
- A Xayaphoummine,T Bucher, H Isambert.Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots.Nucleic Acids Res. 2005;33(Web Server issue):W605-10
- Stanislav Bellaousov, Jessica S Reuter, Matthew G Seetin,David H Mathews.RNAstructure: Web servers for RNA secondary structure prediction and analysis.Nucleic Acids Res. 2013 Jul;41(Web Server issue):W471-4.
- Qiu-Shi Li, Yao-Gen Shu.A critical survey on the kinetic assays of DNA polymerase fidelity from a new theoretical perspective.bioRxiv
- José A. Morin, Francisco J. Cao,J osé M.Lázaro, et al.Mechano-chemical kinetics of DNA replication: identification of the translocation step of a replicative DNA polymerase.Nucleic Acids Res.2015;43(7):3643–3652.
- Robert J. Bauer, Michael T. Begley, and Michael A. Trakselis,Kinetics and Fidelity of Polymerization by DNA Polymerase III from Sulfolobus solfataricus,Biochemistry. 2012; 51(9): 1996–2007.
- J Saturno, L Blanco, M Salas, J A Esteban.A novel kinetic analysis to calculate nucleotide affinity of proofreading DNA polymerases. Application to phi 29 DNA polymerase fidelity mutants.J Biol Chem. 1995;270(52):31235-43.
- C Garmendia.et al.The bacteriophage phi 29 DNA polymerase, a proofreading enzyme.J Biol Chem. 1992;267(4):2594-9.