
Efficient attack on algae.
To conduct an efficient attack on cyanobacteria, we choose to load the peptide onto and into the exosomes so that when it targets the cell membrane, the local concentration of the peptide can be higher than simply diffusing the drug evenly in the water. In this project, the nano-scale exosomes were produced and separated from the engineering E. coli DH5a strain. To promote the production of the exosome, the degP gene in the bacteria genome was knocked out (Schwechheimer & Kuehn, 2013). We applied a two plasmid CRISPR-cas9 system invented by Jiang et al. (2015). First, the pCAS plasmid was transformed into the bacteria through electroporation. Then, positive transformants were selected with kanamycin and made into competent cells. Then, pTagetF-degP and homologous sequences were transformed into the bacteria through electroporation. The positive transformants were selected with Streptomycin. After recovering at 37˚C, two plasmids were cured. Sanger sequencing targeting degP was performed to test whether the gene knock-out was successful. For the gene knock-out strains, they were cultured in M9 media with 2% glucose overnight before harvesting. The bacteria were filtered from the media and then the exosome was purified via ultracentrifugation. To examine the quality and the yield, flow-cytometry and fluorescent staining were performed.
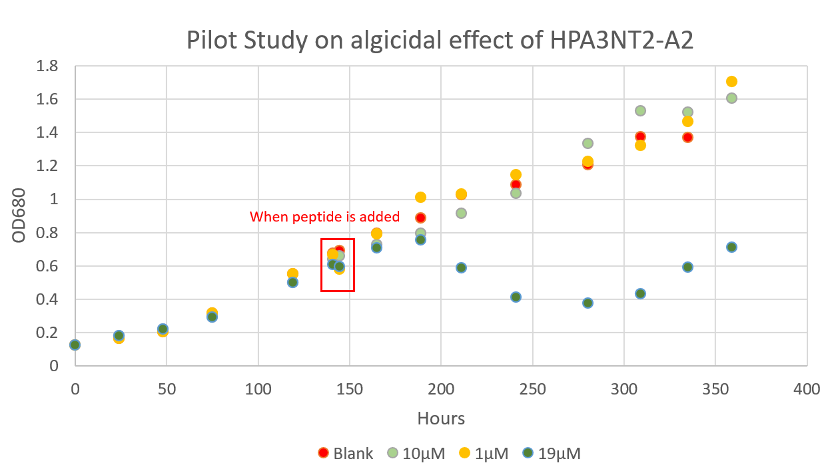
Since the biological synthesis of peptide took a relatively long period of time, we bought chemically synthesized HPA3NT3-A2 to prove its algicidal effects and test its optimal algicidal concentration. Microcystis aeruginosa PCC7806 were cultured in BG11 at room temperature with 40 μmol photons /(m2·sec) light 12 hour every 24 hours and shaking (Fig. 3). Before adding the drug, we measured the whole growth curve of Microcystis aeruginosa to understand its growth pattern and to determine the OD680 of its early exponential growth. A pilot experiment was also done by adding 1, 10 and 19μM of peptide directly into the T25 flask, where the algae was cultured. The pilot study showed that the minimum concentration to control algal growth is between 10-19μM (Fig.4, Fig 5 ). Then, we cultured Microcystis aeruginosa in a cell culture 24-well plate. A concentration of 0 (blank), 15, 20, 30, 40, and 50 μM of peptide was tested, each with 2 replicas. The optical density of each culture (OD680) was tested in the following 5 days to find out the optimal algicidal concentration.


In order to understand the effect of HPA3NT3-A2 on other algae, we streaked our sample taken from Taihu on BG11 plates. After three rounds of streaking, single colonies were obtained, and 9 of them were chosen for strain identification. For strain identification, we conducted 16S rDNA sequencing. Using the 27F & 1492R primer pair, 16S rDNA from all bacterial isolates was amplified by the DNA polymerase master mix . The temperature profile for PCR consisted of an initial incubation at 100 °C for 5 min, followed by 32 cycles of 94 °C for 30 s, 55 °C for 1 min and 72 °C for 2 min, and a final extension step at 72 °C for 20 min. The amplified product was cleaned by agarose gel electrophoresis followed by PCR product purification. Purified PCR products were sequenced and blast with the 16s rDNA database. We will then culture the identified species in liquid BG11 and test the effect of HPA3NT3-A2 on them.
Delivery system
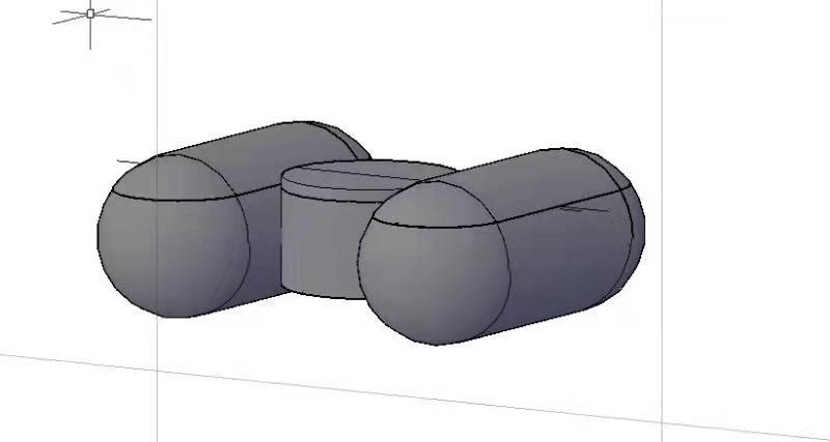
We aim to design a drug delivery system that can slowly release the peptide into the surrounding water. The drug delivery system is composed of small floating units made of biodegradable environmentally friendly plastic called polyhydroxyalkanoates (PHA) whose biodegradation in marine water and the freshwater system have been confirmed (Morohoshi et al., 2018; Sun et al., 2021). Each floating unit consists of two horizontal hollow capsule-like structures to maintain buoyancy and a short vertical hollow cylinder in between to control the sustained release of the peptide. Inside the vertical cylinder, a drug pump is set up in which the peptide and the water-absorbing material are separately wrapped by semi-permeable membrane sacs. Once adequate water flows into the cylinder through a small hole at its bottom, the water-absorbing material starts to expand and squeeze the peptide out and release it into the water system. Multiple floating units can be connected with PHA threads into a bigger net-like unit to flow with the current along with water bloom cyanobacterial species. In this way, the drug is enriched at the water surface to better target the Microcystis aeruginosa.
© Copyright DKU iGEM. All Rights Reserved