Team:Vilnius-Lithuania/Design
DESIGN

Prevention
Usually Entamoeba histolytica infection is treated by metronidazole, which efficiently reduces anaerobic or microaerophilic microorganisms viability. Metronidazole selectivity occurs due to the redox potential of earlier mentioned microorganisms' electron transport components, which are responsible for nitro group reduction. This way, during metronidazole metabolization toxic metabolites such as N-(2-hydroxyethyl) oxamic acid and acetamide in anaerobic or microaerophilic microorganisms are generated. These compounds react with DNA, form adducts with guanosine, and cause cytotoxicity [1]. However, due to acquired resistance to this medicine, higher doses are required [2] and side effects such as nausea, abdominal pain, diarrhea, or in rare cases serious neurotoxicity, optic neuropathy, peripheral neuropathy, and encephalopathy might occur [1]. For this reason, there is a high demand for natural, nontoxic compounds efficiently reducing E. histolytica viability. By following that idea, we did research on alternatives that might be implemented for amebiasis treatment or prevention. During our Integrated human practices and consultation with pharmacologist J. Gulbinovič, we learned that liver abscess forms capsule which serves as a barrier for many therapeutic compounds, so metronidazole is very effective as it can easily cross connective tissue barriers. For this reason, we have focused on amebiasis prevention as currently there is no active prevention of this deadly disease. Despite that amebiasis has been known for more than 160 years, no effective vaccine has come to the market. Furthermore, infectologist A. Marcinkutė stressed that exposure to E. histolytica trophozoites can easily lead to repeated intestinal or even extraintestinal amebiasis (such as an amebic liver abscess). This leads to the inference that immunity can not efficiently fight this pathogen alone - it might need some help from external factors. Read more about our Integrated human practices consultations here.
Naringenin producing probiotics
Why naringenin?
Here comes the idea of naringenin producing probiotic bacteria. Naringenin (Fig. 1) is a natural flavanone produced by fruits (grapefruits, oranges) or vegetables. Recently it has been used for the treatments of cancer, osteoporosis, and cardiovascular diseases, also it has shown lipid-lowering and insulin-like properties [3]. Furthermore, naringenin is effective in controlling several inflammation-related diseases such as sepsis by modulating acute and chronic inflammatory responses by inhibiting pro-inflammatory signaling pathways [4]. Importantly, several studies have reported that naringenin and some other flavonoids have amebicidal activity [5, 6] and naringenin is among the strongest amebicidic potential having flavonoids with its IC50 = 28.86 µg/mL [6]. The exact mechanism of action against E. histolytica is not known. However, there were some ongoing studies of this topic and it will be revealed in future. In addition, the production of naringenin is less complicated than some other flavonoids as it requires only four additional enzymes (Fig. 2), this prevents putting a heavy synthesis burden on the host cell. Finally, optimal intestinal concentrations of naringenin would not only combat amebiasis but also lower the risk of other inflammations in the intestine [4].

Idea of probiotics
As E. histolytica trophozoites grow mainly at the end of the small intestine and encyst in the large intestine [1, 4]; it was essential to prevent infection in both regions comprehensively. The primary prevention idea was to introduce naringenin to the intestine where the amebiasis infection may start and prolong E. histolytica incubation with this therapeutic compound [7]. For this reason, probiotic bacteria were selected as a chassis for the engineered naringenin biosynthesis pathway. Such bacteria are adapted to the harsh intestine environment as it is their natural habitat. To have more abilities if some part of planned experiments would fail, two probiotic strains were selected, namely, Escherichia coli Nissle 1917 (EcN), which are present in the large intestine [5], and Lactobacillus paracasei BL23, which colonize the small intestine [2]. What is more, both of these strains have some additional valuable properties such as anti-inflammatory effects, antagonistic activity against foreign microbes, etc. [3, 6].
Selection of enzymes for naringenin synthesis pathway
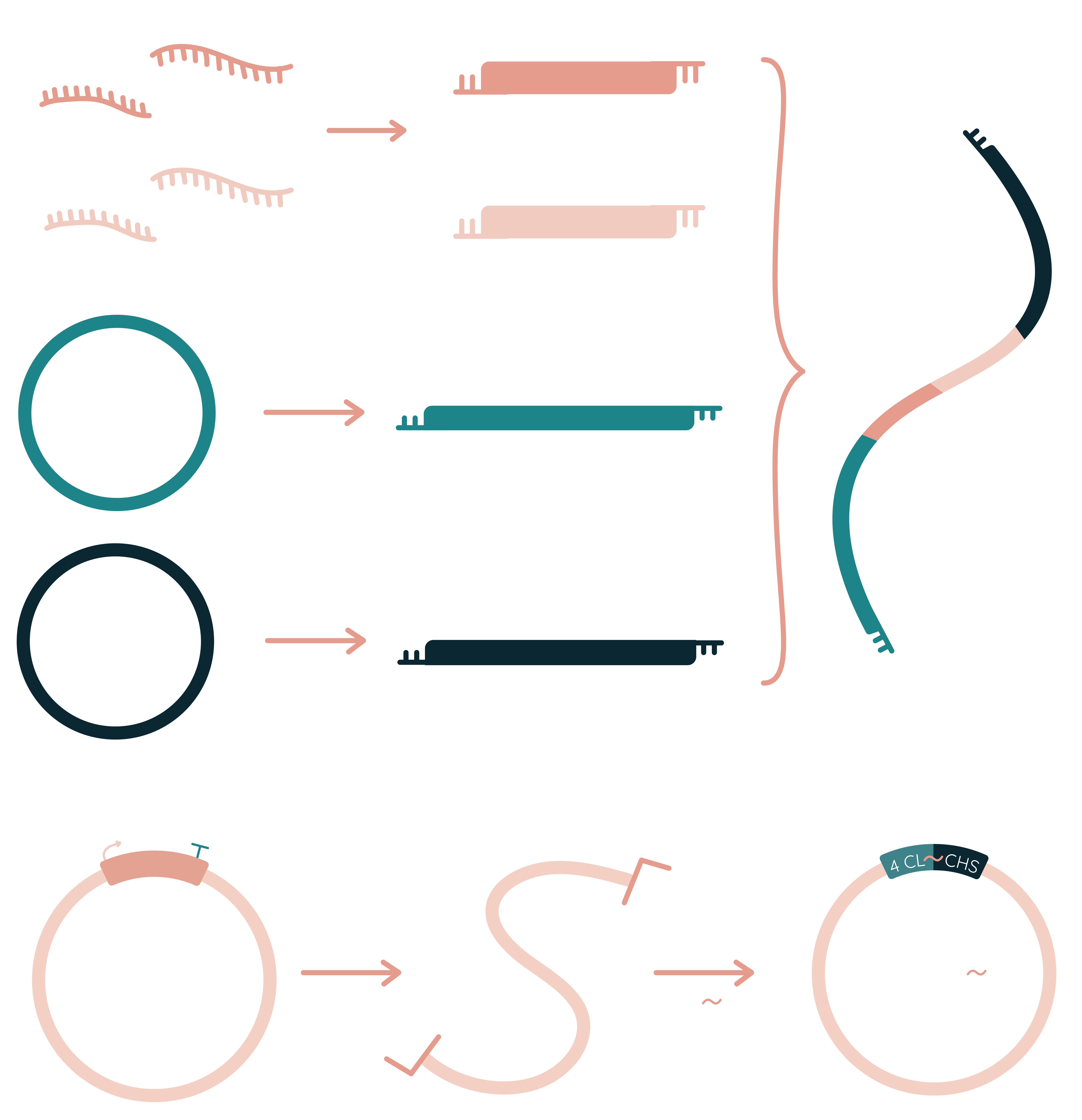
To find the most efficient way of naringenin biosynthesis we had to find enzymes of maximum activity and use them to construct naringenin production pathway in our target probiotic bacteria (Fig. 2). To do so we relied on the article written by Mark S. Dunstan and colleagues [58] and decided to use tyrosine ammonia lyase (TAL) from Herpetosiphon aurantiacus, 4-coumarate-CoA ligase (4CL) from Glycine max, chalcone synthase (CHS) from Arabidopsis thaliana, and chalcone isomerase (CHI) from Medicago sativa. In addition to selecting the most promising enzymes, we also performed codon optimization on the gene sequences of interest.

Codon optimization
We chose two organisms for our naringenin synthesis system - Escherichia coli Nissle 1917 and Lactobacillus paracasei BL23. Nissle is a favorite choice as a gram-negative bacterium for genetically engineered probiotics and L. paracasei BL23 is widely found in many dairy products. Both organisms do not share the same codon frequency, but using the shuttle vector for protein synthesis calls for optimal calculations for protein synthesis.
Since we attempted to optimize proteins needed for naringenin synthesis for two organisms E. coli and L.paracasei BL23, we used chimera-evolve [9] application to optimize sequences for these two organisms. In this context, optimization means minimizing the number of rare codons in both organisms to get the same amino acid sequence. The application performs a genetic algorithm that generates random candidates, scores them, and based on the scores performs tournament selection for optimization rounds. The highest scored sequence is provided as an output. The optimization approach is based on introducing crossovers and mutations, after which the sequences are scored again.
For input, the program takes in coding sequences of organisms that are the target for optimization and the nucleic acid sequences that code the proteins of interest. Since the program did not take any coding sequences (CDS) of E. coli Nissle 1917 genome, we took the E. coli K12 CDS, which is labeled as the reference one. For L. paracasei we picked the CDS of ATCC 393 strain since it was the one that we managed to successfully run on the program. This combination of CDS was used to optimize all 4 proteins of naringenin synthesis. Due to the fact that the program is based on randomization, the optimization was run five times for each protein and the nucleic acid sequences with the highest scores were taken for gene synthesis. Based on the consistency results presented in the paper of the program, the optimal construct scored 6.6306. Although optimization scores of around 3.5 are considered as average, yet these output sequences have a higher score than the sequences designed individually for the organisms taken as targets for optimization. Therefore, the scores that we received were satisfactory to use in our designed system.
| Organism | Used CDS (NCBI Accession Numbers) |
|---|---|
| Escherichia coli K12 | NC_000913 |
| Lactobacillus paracasei | AP012544 |
This codon optimization approach was also used for the VapXD kill-switch system. VapX and VapD coding sequences were also optimized for E. coli and L. paracasei, although this time CDS of BL23 strain was taken as one of CDS inputs for the program.
| Protein | Score |
|---|---|
| TAL | 3.5217 |
| 4CL | 3.5178 |
| CHS | 3.5418 |
| CHI | 3.5991 |
| Organism | Used CDS (NCBI Accession Numbers) |
|---|---|
| Escherichia coli K12 | NC_000913 |
| Lactobacillus paracasei BL23 | FM177140 |
| Protein | Score |
|---|---|
| VapX | 3.5714 |
| VapD | 3.5652 |
Promoter selection and characterization
As in the end, our probiotic bacteria will need to synthesize naringenin under a harsh intestine environment; we wanted to ensure production efficiency as high as possible. For the achievement of this goal, strong constitutive promoters were in need. As experiments with two chassis (Escherichia coli Nissle 1917 and Lactobacillusparacasei BL23) were planned in parallel, during promoter selection process we knew Anderson promoter’s collection to be the best characterized for the E. coli. However, in Lactobacillus sp. case, deep literature analysis was done to select the most appropriate variants.
The idea of sfGFP usage
Life science experiments require many different techniques to visualize intracellular structures or even measure the level of gene expression to characterize parts [10]. For the measurement of chosen promoters’ strength [11] in selected chassis organisms, we have chosen to use superfolder green fluorescent protein sfGPF (BBa_K1365020). This variant of GFP is known as an improved folding protein compared to regular GFP. It allows fusing sfGFP to any protein for expression measurements because it can still gain native tertiary structure despite linked protein folding capabilities. It helps to generate more homogenous fluorescence signals and obtain a more accurate gene expression evaluation. This feature is crucial for measuring proteins of long amino acid chains - tyrosine ammonia-lyase (TAL) or linked 4-coumarate-CoA ligase (4CL) and chalcone synthase (CHS) proteins. Another important note is that sfGFP is more resistant to environmental conditions (pH level) [12], which cause fluorescence fluctuations; unevenness should be diminished or decreased during the fluorescence intensity measurement.
mRNA cyclization system evaluation
Additionally, as a protein production enhancement, we have analyzed a synthetic protein quality control (ProQC) system that should enable translation only when both ends of mRNAs are present, followed by circularization based on sequence-specific RNA–RNA hybridization. As the protein-coding gene is transcribed, its 5’ region forms the hairpin structure in the RBS region. It inhibits ribosome binding and translation initiation if transcription has not ended or the transcript has the truncated 3’ region. This way, RNA cyclization prevents incomplete protein synthesis, wasting cellular resources, and improves full-length protein amount in a cell. The primary idea was to use this system for naringenin synthesis efficiency improvement universally. Recently ProQC has shown production by ensuring the full-length expression of enzymes in biosynthetic pathways, resulting in 1.6- to 2.3-fold greater biochemical production [13].
Firstly, the characterization of the system (BBa_K3904217) was performed by using aforementioned sfGFP protein.

Then measurements were repeated while using longer sequence protein (in particular, TAL) fused with sfGFP.

Finally, the bacteria fluorescence in the presence of the pTRKH2-new MCS with ProQC and without it was compared.
Promoter selection for the final naringenin synthesis construct
The promoters for each component in the naringenin synthesis were chosen according to the information in the article written by M. S. Dunstan and colleagues [14]. It was demonstrated that CHS protein is responsible for the bottleneck reaction in the synthesis chain. Therefore CHS required the strongest possible promoter, as the expression of other proteins - TAL, 4CL, and CHI - could be controlled by the promoter of a weaker expression.
Metabolic pathway construction
To construct the naringenin metabolic pathway firstly we had to construct promoter (selected according promoter strength evaluation results) - gene - terminator systems for each of the enzyme-encoding sequences. This was done by ligation of sticky ends created by specific restriction enzymes. Afterwards, constructed plasmids can be used for full naringenin metabolic pathway creation.
First strategy to construct a full naringenin metabolic pathway was to amplify the pTRKH2 vector and all four naringenin synthesis gene cassettes using primers that contain specific restriction endonuclease recognition sites. This way we should be able to digest each sequence with appropriate restriction enzymes and create a library of inserts with sticky ends that can be ligated into the target vector as the ending part of composite insert or as a part of the whole naringenin synthesis cassette.

Another strategy that we chose for reaching this goal is Gibson assembly. By amplifying the pTRKH2 vector and all naringenin synthesis genes with primers containing flanking regions that form homologous pairs with each other in the manner that a complete naringenin synthesis cassette can be constructed in a single tube reaction.

Lastly, after a broad literature analysis we have found that some flavonoids synthesis is more efficient then required metabolic pathway is divided into two separate bacteria which are grown as cocultures [24, 59, 60]. This strategy lowers the metabolic burden loaded per cell. Keeping this in mind, we have approached the third strategy - coculture of the divided metabolic pathway into three parts (fig. 8). Moreover, based on literature analysis [21, 22, 23] we have chosen to modulate the inner metabolism of our chassis probiotic. For this purpose we have chosen to knockout 4 different genes - ackA, pta, adhE, tyrP (more about these genes encoding proteins biological importance you can find in the paragraph of genome editing). Desired coculture structure:
- tyrP knockout E. coli Nissle 1917 harboring TAL encoding gene. tyrP knockout should increase endogenous L-tyrosine concentration which is the substrate to the first enzyme of the naringenin production metabolic pathway.
- ackA, pta and adhE triple knockout E. coli Nissle 1917 harboring linked 4CL and CHI encoding genes. ackA, pta and adhE triple knockout leads to increased malonyl-CoA concentration. This is a particularly important improvement in the bottleneck reaction of naringenin synthesis.
- wilde type E. coli Nissle 1917 harboring CHI encoding gene. As CHI substrat is only CHS produced naringenin chalcone, no additional endogenous metabolism modifications would improve this reaction. Therefore, no genetic manipulations is needed in this region.

Construction of pTRKH2 vector with new MCS
For the construction of the naringenin synthesis cassette that would effectively work in both Escherichia coli Nissle 1917 and Lactobacillus paracasei BL23, we chose the pTRKH2 shuttle vector. This plasmid is suitable for E. coli and gram-positive bacteria, with a high copy number in gram positives [8].
However, to construct a pTRKH2 vector that would fit all our desired inserts, we needed to modify it by changing the plasmid’s original multi-cloning site, since the original pIC-19R multi-cloning site has been found to contain restriction sites that were not suitable for our desired genetic manipulations.
The new multi-cloning site was designed in a way that would grant easy genetic manipulations of this vector. By manipulating different restriction enzyme recognition sites, we can insert and remove promoter sequences, mRNA cyclization system, and genes encoding for naringenin synthesis proteins. This vector construction strategy allows us to construct all the vectors we need in a similar manner and evaluate the efficiency of promoters of interest as well as composite constructs in order to select the most promising sequences for the development of an effective probiotic strain.
We constructed the designed sequence by annealing together four oligos and this way creating sticky ends needed for the insertion of the new multi-cloning site into the pTRKH2 vector.
To construct a pTRKH2 vector with a changed multi-cloning site, we eliminated the unnecessary sequence by amplifying the pTRKH2 sequence with PCR primers that would create the deletion of this specific region in the plasmid.
The amplified linear pTRKH2 vector was digested with appropriate restriction enzymes and ligated together with the new multi-cloning site sequence.

Kill-switch
To prevent our genetically modified probiotic bacteria from escaping the intestine to the environment, they were provided with the disabling mechanism, which could be activated after exposure to some environmental factors. This goal was obtained by implementation of the newly described type II toxin-antitoxin (TA) system VapXD from non-typeable Haemophilus influenzae [15]. A recent publication showed that it consists of homodimer toxin VapD and a single antitoxin VapX. For the neutralization of the toxin, one antitoxin binds two toxins in an unusual stoichiometry 2:1. What is more, antitoxins must be continuously synthesized to ensure bacteria survival.
In the context of our project, we wanted to design the kill-switch regulated by bile salts and temperature. This choice can be justified by the difference of bile salts concentrations and temperature inside and outside of the intestine. As seen in the figure 11, our system consists of bile-inducible promoter (BBa_K3904002), VapX antitoxin (BBa_K3904001), cold-inducible promoter (BBa_K3904003) and VapD toxin (BBa_K3904000).

If probiotic bacteria are present in the intestine where bile-salts occur, and temperature is around 37 °C, bile-regulated promoter induces antitoxin production, as cold-inducible promoter activity is inhibited. However, if bacteria escape, a bile-inducible promoter is inhibited, as cold-regulated promoter causes greater toxin production and lowers bacteria’s chance of survival.
Overall construct has two different promoters but the same RBS, so production can be altered by changing promoter intensity. This way, future teams could characterize this system more thoroughly and adapt for their projects.
| Part | Name | Description |
|---|---|---|
| BBa_K3904002 | Bile-inducible promoter | Promoter induced by bile-salts, originally from Lactobacillus paracasei BL23. |
| BBa_K3904001 | VapX | A small, monomeric, single-domain antitoxin of the VapXD system. |
| BBa_K3904003 | Cold-inducible promoter | Promoter originally responsible for cold-shock response protein production in E. coli. |
| BBa_K3904000 | VapD | The toxin of the VapXD system. It is an endoribonuclease known to be active as a dimer. |
VapXD comparison to MazEF
Usually, to prevent genetically modified bacteria from spreading to the environment, the mazEF toxin-antitoxin system is used. VapXD and mazEF work similarly, as overproduced antitoxin binds to the toxin and inhibits its endoribunucleatic activity [16, 17]. However, as seen in table X, VapXD kill-switch consists of shorter sequences. This point becomes beneficial when the toxin-antitoxin system is needed to be inserted in the presence of bigger genetic constructs. From this lower metabolic burden arises and ensures higher genetic manipulation efficiency [18].
| MazEF | VapXD | |
|---|---|---|
| Protein length | MazE* - 89 aa
MazE** - 82 aa MazF* - 119 aa MazF** - 111 aa |
VapX - 63 aa
VapD - 92 aa |
| Activity | Endoriboncleatic | Endoriboncleatic |
| TA type | II | II |
* Toxin-antitoxin system, Atu0939 (mazE-at) and Atu0940 (mazF-at), in the chromosome of Agrobacterium tumefaciens.
** Toxin-antitoxin system from Escherichia coli (strain K12).
Protein scaffold
As for another enzyme catalytic efficiency enhancement, protein scaffolds have great opportunity being the perfect solution [19]. Gathering enzymatic reactions into one place by binding to nucleic acids or proteins could speed-up limiting rate reaction steps [20]. One way to do this is by attaching enzymes to DNA/RNA with fused zinc fingers.

In previous works by many scientists we found information about proteins that bind plasmid DNA, but we thought this is not enough for making in vivo reactors. This year we focused on doing In vivo reactions. They let us overcome expensive cofactor needs and are rapidly developed.
We created a RNA scaffold to which DNA/RNA binding proteins can bind.
Created 3D models:


This structure folds into optimal tertiary structure for further protein binding. However it is vital to make two loops without Ts as it would create transcription termination points. Used sequence:

Secondary structure:

Stems show potential for further sequence increment if needed. The only problem is transcription which could terminate early or could not synthesize this RNA at all.
However we did not finish experimental trials with this kind of design and mainly focused on creating fusion proteins as they are more promising.
Fusion proteins
Based on the literature analysis [51, 52, 53], we found out that beneficial interaction between two enzymes takes place after incorporating linkers that are composed of 5 to 15 amino acid residues. We included GSG linker because 4CL from Arabidopsis thaliana - a homolog to the protein of our choice - has already been linked with stilbene synthase (STS). There were in vitro kinetic assays performed for the 4CL:GSG:STS complex and its crystallographic structure (PDB: 3TSY) was deposited to Protein Data Bank. In summary, we chose to connect our proteins using linkers that are flexible: GSG, GGGGS, (GGGGS)2, (GGGGS)3; and rigid: EAAAK, (EAAAK)2, (EAAAK)3.
More about Fusion protein model.
The whole construction idea was to insert restriction sites in a protein encoding sequence that could help us construct flexible and rigid linkers between 4-coumarate-CoA ligase 2 (4CL) and chalcone synthase (CHS). We were interested in fusing two proteins without inserting any additional amino acids to C and N protein terminus. In this manner, we designed oligonucleotides that would be annealed and used as linkers between coding sequences in fusion protein cloning.


Genome editing
To assure effective and consistent naringenin synthesis, we chose to insert the cassette of genes responsible for the probiotic synthesis into the genomes of Escherichia coli Nissle 1917 and Lactobacillus paracasei BL23. CRISPR-Cas9 and Lambda Red gene recombination (fig. 20) or CRISPR-Cas9D10A nickase-assisted genome editing (fig. 21) technologies have been applied to perform this genetic manipulation. The main differences between these two systems:
- Differently than E. coli Nissle 1917, L. paracasei BL23 has poor capability to repair double strand DNA breaks. For this reason, instead of regular Cas9 endonuclease, used for E. coli Nissle 1917 genome editing, Cas9D10A nicase is used for L. paracasei BL23 genome editing.
- L. paracasei BL23 genome editing relies on endogenous recombination machinery while for E. coli Nissle 1917 genome editing exogenous Lambda Red recombination system is engaged.
- Donor DNA for homologous recombination in E. coli Nissle 1917 is double stranded linear DNA fragment which is introduced into cells by co-electroporation together with pTarget plasmid. Meanwhile, donor DNA for homologous recombination in L. paracasei BL23 is a part of pLCNICK plasmid.


Firstly, we needed to decide what regions in the genomes of our probiotic strains would be the most suitable for the insertion of the naringenin synthesis cassette. The ideal place in the genome would be a sequence, where insertion of a foreign DNA would not result in the disruption of genes necessary for cell viability, and naringenin synthesis would take place as efficiently as possible. For that reason, we have made broad research on probable insertion sites. We took into account the possible transcription level based on nearly located genes or overall genomic locus [25] and chosen gene function. This led us to nupG and putative colicin encoding gene. NupG is a nucleoside permease coding gene in E. coli and putative colicin gene is probably encoding for colicin - a type of bacteriocin produced by and toxic to some strains of E. coli. These genes have been chosen for the metabolic pathway insertion because of probably high transcriptional potential in this region and minimal damage to the host [26].
Secondly, it is important not only to integrate the metabolic pathway cassette, but also modulate the host metabolism toward the more efficient naringenin production. For this reason we chose to knockout several E. coli Nissle 1917 genes coding enzymes related to the malonyl-CoA metabolism or tyrosin transport. Acetyl-CoA serves as the first flux control point for flavonoid biosynthesis, whereas malonyl-CoA serves as a starting point for the synthesis of flavonoids, which are only consumed for synthesizing fatty acids. To channel carbon flows toward acetyl-CoA, the availability of acetyl-CoA precursors such as pyruvate needs to be increased, or the consumption of acetyl-CoA in other central metabolic pathways, such as the tricarboxylic acid cycle and glycolysis, needs to be reduced [23]. Having the aforementioned criteria in mind, we selected several potentially suitable gens in the E. coli Nissle 1917 genome for the knock-out in order to enhance the efficiency of naringenin synthesis machinery:
AckA-pta operon. Disruption of this genomic region is known to limit the acetate formation from acetyl-CoA, increasing the cellular concentration of acetyl-CoA up to 16 percent [21]. Increased concentration of this molecule theoretically should result in enhanced malonyl-CoA formation and consequently more effective naringenin synthesis since the amount of malonyl-CoA available in the cell is the limiting step of naringenin production [23].
Acetaldehyde dehydrogenase (adhE) gene. Knockout of adhE gene disrupts the conversion of acetyl-CoA to ethanol in the cell, therefore more acetyl-CoA can be converted to malonyl-CoA. This molecule is used by chalcone synthase during the production of naringenin chalcone, which is later converted to naringenin [23].
tyrP gene is known to be involved in transporting tyrosine across the cytoplasmic membrane, therefore tyrP knockout mutants are potentially able to produce 10 percent higher amounts of L-tyrosine than that of the original strains [22]. This L-tyrosine then can be converted to p-coumaric acid by tyrosine ammonia-lyase (TAL), which is further utilized to make p-coumaroyl-CoA by 4-coumarate-CoA ligase. The sequential condensation of one p-coumaroyl-CoA and three malonyl-CoA leads to the formation of naringenin chalcone by chalcone synthase (CHS), which is then converted to naringenin by chalcone isomerase (CHI) [24].
Since we were not able to obtain fully reliable Lactobacillus paracasei BL23 genome annotation, we decided to rely on the experience of our advisors and based on their consultations, insert the sequences encoding for naringenin synthesis machinery to one already known suitable place in the Lactobacillus paracasei BL23 genome using pLCNICK vector, which is designed with the purpose of genome editing for Lactobacillus paracasei BL23.
Naringenin synthesis evaluation
In order to confirm that we synthesized Naringenin we chose the HPLC-MS method. Two options were selected as viable solutions finding it - directly from medium or by extraction with ethyl acetate. For the first technique time consuming preparation is needed but it ensures that all main compounds are still intact in the sample. Alternatively extraction from medium is far better for whole HPLC-MS equipment however more components can be lost during this process due to higher solubility in ethyl acetate.

Diagnostics
Currently, detection of Entamoeba histolytica relies on inaccurate microscopy methods as well as expensive and time-consuming ELISA techniques [54]. Since these approaches require a lot of resources, time, and experienced personnel, they are insufficient in quickly and accurately diagnosing millions of people affected by this disease every year. An untreated infection, in some cases, can migrate through the whole organism and reach liver, lungs, or even brain. Aside from an effective prevention system, a clear solution to this problem could be early detection of potentially dangerous infection. This can only be done by developing an inexpensive, rapid, and user-friendly test for the identification of Entamoeba histolytica infection. To reach this goal we decided to apply the systematic evolution of ligands by exponential enrichment (SELEX) method, which is a technique for producing oligonucleotides known as aptamers that specifically bind to target molecules (Fig. 23) [55]. By implying this method we decided to develop an aptamer-based Entamoeba histolytica identification system that is based on target protein detection using DNA aptamers. As potential target, we chose pyruvate phosphate dikinase (EhPPDK) - a metabolism protein of Entamoeba histolytica that is found in infected blood [56], and cysteine proteinase 5 (EhCP5) - an enzyme that is used by the parasite to go through the human intestine walls [57]. After evolving our aptamers to detect either of these specific proteins, we can conjugate it to polydiacetylene (PDA), which can form nanostructures that can potentially generate a colorimetric response [47]. Our test is created to change its color from blue to red when aptamer captures the target biomarker and indicates Entamoeba histolytica infection.

EhPPDK and EhCP5 synthesis
In the diagnostic kit creation part, we firstly needed to obtain functional Entamoeba histolytica biomarkers. For this purpose we chose two proteins secreted by the ameba: pyruvate phosphate dikinase (PPDK) [48, 49] and cysteine proteinase 5 (CP5) [50] that distinguish our target organism from other amebas. We have chosen to use E. coli BL21 strain designed for enhanced recombinant protein synthesis. For protein expression, we modified the pET28a+ vector. Originally, this plasmid contains N-His and C-His tags and the space between N-His-tag and protein contains several random nucleotides and thrombin recognition sites. For this reason, this original vector is not fully suitable for further PPDK or CP5 usage in SELEX, since we need to acquire the native form of protein in order to collect aptamers specific to PPDK or CP5 and a short sequence of random amino acids might cause a selection of nonspecific aptamer. What is more, we also needed to expose the largest amount of protein surface to aptamers to enhance the possibility of acquiring the best variant of specific aptamers. To do that, we chose to modify pET28a+ by removing the unwanted N-His tag together with the thrombin recognition site. Instead of aforementioned unwanted sequences we inserted a RBS cassette to acquire only C-His tag containing plasmid. Also, we needed protein with N-His tag and native form of it, therefore we inserted RBS plus N-His or N-His-Tev cassettes which enabled us to clone PPDK or CP5 gene with N-His tag or N-His plus TEV protease recognition site tags. We decided to choose TEV protease instead of thrombin, since TEV protease is by far the best studied and most widely used potyviral protease. TEV recognition site is required to obtain native PPDK to measure aptamer specificity and for this reason His tag on the C or N end of the protein might cause some false-positive results as it also could interact with ssDNA aptamers.
His-tag plasmids usage justification
AmeBye detection test creation requires synthesizing and purifying recombinant Entamoeba histolytica proteins that could be later used in aptamer evolution. There are several protein purification strategies (Dialysis, Gel-Filtration Chromatography, Ion-Exchange Chromatography, Electrophoresis, Affinity Chromatography, etc.) [27]. In our case, we have chosen to use an Immobilized Metal-Ion Affinity Chromatography because it generates acceptable results - only one required purification step in order to get relatively pure protein. In particular, we have chosen to produce recombinant proteins with 6 histidine residues tag at the C or N-terminal of the protein. This moiety interacts with cobalt or nickel ions, which also creates coordinative bonds with nitrilotriacetic acid attached to the chromatographic matrix. This way, a recombinant protein with His-tag binds to the chromatographic column, while other proteins, without His-tag, are washed out. The competitive agent - imidazole - displaces specific protein and, later on, it is eluted [28]. This technique simplifies purification, reduces the number of chromatographic steps. His-tagged protein could be obtained through a single purification step and the His-tag might be removed by specific protease [29]. Recombinant E. histolytica proteins with His-tag were needed for SELEX experiments, where we immobilized proteins on the silica beads. Those beads’ surface is derivatized with the nitrilotriacetic acid (NTA) chelation moiety and loaded with divalent nickel ions (Ni+2). It enables the capture of proteins and, in our case, evolutionize ssDNA aptamers affined to them [30, 31].
For cloning, we have used pET28a(+) plasmids and their modified versions: pET28-N-His (BBa_K3904318), pET28-N-His-TEV (BBa_K3904319) and pET28-C-His (BBa_K3904320). Modifications have been done in our laboratory in order to get vectors with only N- or C-His tags or N-His tag and TEV recognition site between N-His tag and protein-coding sequence. TEV sequence is needed to get native protein after purification. N and C-His tags have been chosen because we decided to make sure that all sides of EhPPDK and EhCP5 would be exposed to the solution where ssDNA aptamers would bind to them. This in vitro aptamers evolution strategy should generate a higher number of candidate aptamers that have been screened by NGS and obtained results examined. We have chosen a strategy to obtain a large number of aptamers because we can not determine where aptamers would specifically bind to the protein, so we needed to enlarge the exposed protein surface.
Aptamers based diagnostic test
Right now, the only methods to detect the infection are based on microscopy or antibodies. While ELISA is accurate, it also needs experienced and qualified personnel, expensive equipment and way more time. We chose aptamers as our diagnostic probes because they can bridge this gap and give as the best of both worlds - a fast, inexpensive and reliable method of detecting a biomarker. Aptamers are simple single strands of DNA which makes them inherently more stable and easier to use than protein antibodies in the ELISA assays. Since it is not constricted by the bonds with a second strand it can bend in various ways and bond with itself. The structures are completely random but with a 1015 of unique sequences there's a chance that one of them will contort into a shape which can interact specifically with our target molecule.
Aptamers versus antibodies
| Aptamers | Antibodies | |
|---|---|---|
| Chemical modifications | 5’ and 3’ end modifications | Conjugated with one type of signaling or binding molecule |
| Stability | Stable at room temperature, have longer shelf life. The denaturation is reversible | Sensitive to temperature, require refrigeration. Irreversible denaturation |
| Size | Mainly small molecules 30 to 80 nt (~12 – 30 kDa) that display better access to their target | Large molecules (~150 kDa), resist to filtration by the kidneys |
| Production time and cost | In weeks time | In several months |
| Reproducability | Precisely in chemical manner | From animals not identically |
| Degradation | When exposed to nuclease activity | Have long half-life |
| Chemical modifications | Increase stability and applicability | Hardly customizable |
Generating aptamers
Making aptamers without SELEX
EFBALite
This year we wrote a program EFBALite that generates aptamer for a target protein molecule from scratch. EFBA is an acronym that stands for an entropic fragment-based approach. As the name suggests the algorithm is based on a statistical measure called relative entropy (or Kullback–Leibler divergence). The algorithm of the program is:
- Constructing a discrete space on the surface of a target molecule in order to fix the position of the first nucleotide;
- The first nucleotide is rotated by 0 and 180 degrees along all Euler angles, where O3 atom is considered as the zero point of Euclidean space;
- The nucleotide is chosen the one that has the highest relative entropy;
- An information gain threshold T, which each nucleotide in the sequence must reach, is specified;
- For the subsequent nucleotides, we keep the already determined sequence fixed in space and rotate the next nucleotide by -90, -60, -30, 0, 30, 60 and 90 degrees along all Euler angles by considering the phosphate atom as the zero point of the Euclidean space;
- If the maximum relative entropy is higher than T, keep the nucleotide, fix it to the most probable position and continue from 4th point, if not backtrack to the first nucleotide and check the second most probable position;
- Continue this until you find a sequence of desired length or exhaust all possible sampling positions.
Aptamer docking
In order to validate the generated aptamer sequences in silico, we included an aptamer docking workflow. This flow included following steps:
- Determination of 2D structure of the aptamer sequence with length N using Mfold [40];
- Determination of 3D structure of the aptamer sequence using RNAComposer [41];
- Docking of the aptamer to its target molecule with HDOCK [42];
- Determination of 2D structure of the random aptamer sequence with length N using Mfold;
- Determination of 3D structure of the random aptamer sequence with length N using RNAComposer;
- Docking the random aptamer to the target molecule with HDOCK;
- Comparing HDOCK scores of the random and the generated aptamer.
If the score of the generated aptamer is lower (more negative) than the score of the random aptamer, the generated aptamer is considered as valid for experimental evaluation of the affinity.

Transformers enhanced aptamer design software (TEA)
We took a step further and applied a novel transformer-based neural network (NN) model Albert which was combined with a genetic algorithm to make aptamer generation in silico a faster and more resource-efficient process. Firstly, the algorithm was enhanced with a Bayesian probabilistic model to define a finite GA iteration number that helps users to determine when a list contains a proper number of fit aptamers. Next, employing MCMC methodology we were able to analyse the NN model error rate which gave insights about probability of throwing away the fit aptamer from a final list and space for future improvements. Finally, the Pytorch NN model was rewritten to the ONNX framework which sped up the algorithm more than 3 times, and overall more than 300 compared to EFBALite.
More information can be found about these software tools on Software page.

Making aptamers with SELEX
Making aptamers from scratch requires a method called SELEX - systematic evolution of ligands by exponential enrichment. Both protein and cells (target) [35] can be used in SELEX for generating aptamers. In our case we immobilized our desired protein on magnetic beads and incubated with ssDNA aptamer pool by varying time.

In the initial library there are 1012-1015 different sequences. Each of them have a unique tertiary structure and possibly bind to a specific target. Any unbound sequence has less of a chance to survive washing steps and thus is gone forever and ever. Bound sequences are amplified using PCR with universal primers for all aptamers and used in the next SELEX rounds. It usually takes 8-12 rounds of SELEX to evolutionize affine and selective aptamer structures.
ePCR vs oPCR
After each selection round properly binding aptamers undergo amplification for further selection. One way to do it is by traditional PCR, but after researching short fragment amplification we stumbled upon a method called emulsion PCR (ePCR) [36] in which PCR is done in small unique chambers - micelles. In each 50 µl PCR test tube there are around 109 micelles in all of which PCR happens independently of one another. Each chamber produces DNA from a template inside and after it runs out of all dNTPs no more non-specific fragments are produced as it happens during traditional open PCR (oPCR).
Emulsion PCR (ePCR)
Background
After getting some insights about short fragments amplification we decided to make an emulsion PCR for our aptamers [37]. Compared to oPCR less non-specific products can be obtained after purification [38]. However specific materials are needed for ePCR and lengthy procedures make it less accessible for wider usage.

Emulsion PCR (ePCR) v.1
First method of ePCR is to use materials from microfluidics emulsions such as - Span 80, Triton X-100, Tween 80. They are the simplest compounds widely used in many molecular biology methods, however Triton X-100 is characterized as carcinogenic and thus it should be changed to other alternatives for further research.
Emulsion PCR (ePCR) v.2
Updated composition: 4% ABIL EM 90, 0.055% Triton X-100. These compounds are used in micelles that encapsulate cells and withstand multiple freezing and thawing conditions [39]. For this reason this composition was chosen as an alternative to original v.1 for better performance.
In the end we did not choose ePCR for aptamer amplification. oPCR was a simpler method to optimise quickly in a short matter of time. We hope to introduce this technology for wider applications in iGEM context.
Data analysis. Illumina + Sanger sequencing
Sequencing by two methods - Sanger and New Generation by Illumina provide full information about developed aptamers [43]. The first one is to gather minimal data - by ligation of aptamers from 6th, 8th and 10th cycles with blunt ends to pUC19 and sequencing by Sanger method using M13 primers. The second one enables us to make deep analysis about how each cycle performs and gather information about motif saturation.
After getting Sanger sequencing results .ab1 files were converted to .fasta. From there aptamers were found using primer sequences from SELEX. We included both blunt end ligation cases - forward and reverse. All sequences were normalised by taking into account poor reading scores.
To sequence aptamers by NGS [44, 45] we chose to add specific adapters and indexes ourselves and to use Illumina MiSeq platform as specified in Usage Instructions. Each SELEX round were assigned to Nextera indexes i5 in 5’ end and i7 in 3’ end of DNA. All used indexes including flanking sequences can be found in parts.
PDA synthesis
Ensuring that the diagnostic test has colorimetric value for illness determination is essential in our case as it is not connected to any other external device. To see qualitative values we chose polydiacetylene (PDA) as the sole provider of color transition when aptamers catch biomarkers [46]. It is possible to create two approaches for detection - one based on paper and another by means of liposome technology [47]. We chose the first one because of the easy user interface and ability to withstand environmental changes.
All syntheses were done in our laboratory and scaled up for making sure many parameters are tested for optimal colorimetric response. In (Fig. 28) the main reaction of aptamer conjugation with fatty acid is depicted.

Such activated aptamer is used in further PDA development.

Hardware
Overall, the final product - aptamer-based vertical flow detection device - and all of the technology behind it lies inside of the hardware. In our case it is a 3D printed diagnostic test outter case. It contains 3 oval windows for sample analysis, one for our biomarker (PPDK) and two control windows. As our test is based on colorimetric response, colorimetric assistance is applied to make it more inclusive. If the end-user observes merging of colors, it indicates positive result for amebiasis. If the user is not infected or diagnostic test does not work, at least one of all windows will not have merged colorimetric response with outer ring, meaning that test is negative or failed.
Find out more about the hardware here.
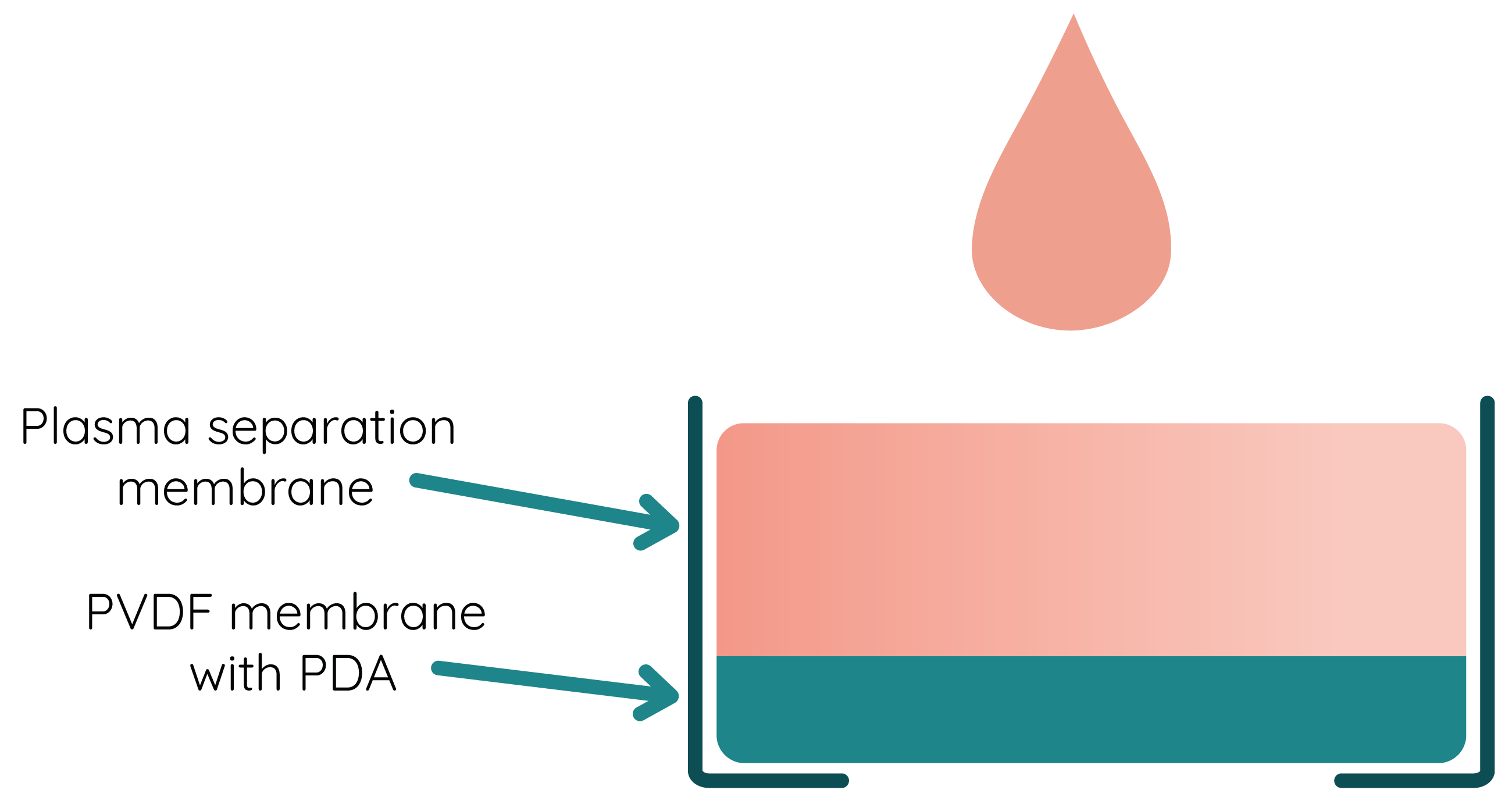
Membranes
Working with viscous liquids like blood requires optimal flow of all molecules. To filter from red blood cells we integrated plasma separation membrane. This results in a high-quality plasma production from whole blood in a few minutes. As blood separation ensures minimal interference of blood cells in the process of detection, it allows to maximize the performance of our test. Moreover, plasma separation adds to improved sensitivity and selectivity of the created test.
The concept behind all of this is asymmetric membrane which should not obstruct flow water with biomarkers. The cellular components of the blood are to be captured in the larger pores of the membrane, while the plasma flows down into the smaller pores on the downstream side of the membrane. The rapid separation process yields plasma similar to traditional centrifuged plasma.