Overview
Homology Modeling of Flap Endonuclease
RAD27 5’ flap endonuclease, nicknamed Flappase in the context of this project, plays a critical role in the removal of a 5’ overhanging substrate in DNA; in vivo this is typically to maintain genomic integrity. Flappase is an integral component of the Invader assay used to detect single nucleotide polymorphisms (SNPs) in the 2021 SUNY Oneonta iGEM project SNflaPs. Much of the literature on the Invader assay does not specify the source of the enzyme or uses human flap endonuclease (FEN1). For our use, Saccharomyces cerevisiae (S. cerevisiae) was chosen as the source of the enzyme due to its practicality in the laboratory. Yeast enzymes typically function well over a wide range of temperatures, making them well-suited for field applications.
There is currently no known crystallographic structure for RAD27, therefore implementation of homology modeling is needed to produce a viable structure that will provide insight into RAD27 function. One intent is to confirm that the assay methods used for FEN1 are transferable to RAD27, predicting detection assay success. Last year, Team SUNY Oneonta produced a rough, non-optimized model of RAD27. Our goal this year was to produce a more refined and validated model (Figure 1). The information contained within this new model will be useful for providing insight on target oligonucleotide recognition and affinity, multiple SNP detection, and steric parameters.

Figure 1: Homology model of S. cerevisiae RAD27 with labeled domains: helix-turn-two-helix (purple), hydrophobic wedge (orange), helical cap acts as active site (red), helical gateway (yellow), DNA (green)
Method
Template Selection
The target sequence of RAD27 was found using UniProt. Through the Basic Local Alignment Search Tool (BLAST) the target sequence was compared to similar protein sequences and filtered out based on the availability of a known structure. The original pool of templates included FEN1 from P. furiosus (PDB ID: 1B43), two Homo sapiens (PDB ID: 5UM9 and 3Q8L), and D. amylolyticus (PDB ID: 3ORY). Statistical data (Figure 2) concluded the most compatible sequence was Homo sapiens FEN1 3Q8L. An alignment between the two sequences allowed for visual representation of amino acid differences (Figure 3).
| BLAST | Quality Data | Ramachandran |
|---|---|---|
| Source Identity: 61.08% | QMEAN: -0.49 | Favored: 96.45% |
| Expected Value: 2.00E-144 | GMQE: 0.79 | Outlier: 0.30% |
Figure 2: Statistical analysis of target versus template sequence
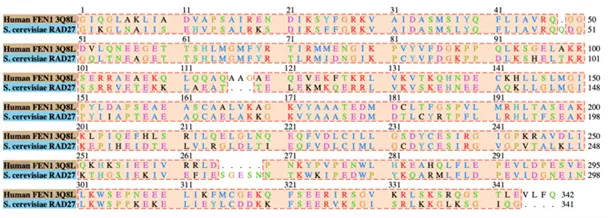
Figure 3: Sequence alignment between target S. cerevisiae and template Human FEN1 emphasizing sequence variation.
Creating the Model and Structural Alignment
The SWISS-Model server was used to produce a homology model by inputting the target sequence (RAD27) and the template (FEN1) PDB file 3Q8L (Figure 4). The resulting Ramachandran plot (Figure 5) depicts the secondary structure of the protein, which fell mostly within allowed parameters. A structural alignment in Chimera (Figure 6) yielded the root mean squared deviation (RMSD) of 0.161Å and provided insight into how similar the model structure is to the template. Additionally, this template structure is bound to DNA which allows visualization of how RAD27 will interact with the designed oligonucleotides.

Figure 4: Homology model of Flappase produced using the target sequence (RAD27) and the template (FEN1) (PDB file 3Q8L). Dimensions of entire protein depicting the capacity for DNA to enter the active site; C and N terminals also labeled (red).

Figure 5: Ramachandran plot of homology model, RMSD 0.161Å

Figure 6: Structural alignment of RAD27 (blue) and FEN1 (red)
Simulation Preparation
To gain computational information on the stability of the RAD27 model and further verify its accuracy, it must be subjected to simulation testing. The AMBER program was used to perform a molecular dynamics simulation. The coordinates for the homology model were prepared for simulation using the CHARMM-GUI server.
Insights Gained From The Model
The goal of the SNflaPs project is to provide a panel of genetic testing in cattle. While our primary test involved a singular SNP, many of the traits in question have multiple SNPs in a gene. It was in our interest to also provide insight as to how Flappase would interact with DNA in cases like this. The first question addressed by our model was what the minimum distance between target sites should be in order to accommodate the flappase protein if we wish to detect multiple SNPs using our system. Based on the dimensions measured in the model (Figure 4), we determined that sites must be 75 Angstroms apart. This corresponds to 22 nucleotides . Our next step was to mutate the DNA sequence in the active site of the template structure to our target sequences (Figure 7) in order to assess possible nucleotide affinity and predict potential steric hindrances.
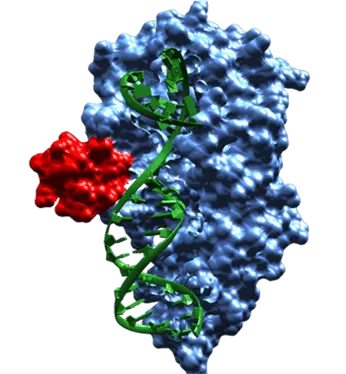
Figure 7: Model showing active site (red)
References
Cordice-Little, B., & Ibrahim, A., iGEM Team 2020, Ca2lf. Team:SUNY oneonta/model. https://2020.igem.org/Team:SUNY_Oneonta/Model.
Tsutakawa, S., et. al., 2011. Human Flap Endonuclease Structures, DNA Double-Base Flipping, and a Unified Understanding of the FEN1 Superfamily. Cell, 145(2), pp.198-211.
Software BLAST, SWISS Model, Chimera, Chimera X, CHARMM-GUI, AMBER20
The SUNY Oneonta iGEM team members would like to thank Dr. Jill Fielhaber and Dr. Kelly Gallagher for guiding us through our project. A special recognition to Dr. Angela Migues for sharing her expertise throughout this aspect of the project.