Team:Chalmers-Gothenburg/Design
Design
The first part of our engineering cycle
SUMMARY
We have designed a tunable Cell Factory based on yeast, that is able to produce a myriad of different fatty acid (FA) profiles in a flexible way. These profiles can be tuned according to the desired profile of the end-product, e.g., to mimic food oils or other industrially relevant lipid formulations. To achieve this, we had to introduce several genomic modifications into our yeast, consisting of three main parts. First, we introduced three vectors containing genes coding for enzymes called thioesterases that can terminate the FA elongation process at different lengths. Each of the three thioesterases have a preferred FA termination length, corresponding to short, medium and long chain FAs. Second, since different end products consist of specific ratios of FA chain lengths, we coupled the expression of each thioesterase to the addition of inducer substances, making the FA profile of the yeast cell tunable. Third, in order to be able to produce the desired FAs we also introduce a new Fatty Acid Synthesis (FAS) system, to the yeast genome. This FAS system originates from E. coli and consists of eight genes. To further improve the FA synthesis, we also tried to downregulate the FAS system native to the yeast using an inhibiting antisense RNA. We will now go through each of these 3 parts in more detail. All designed plasmids and integrated gene fragments were designed using Benchling and ordered from Integrated DNA Technologies and primers were ordered from Eurofins Scientific.
Details about the experimental procedures
can be found in the experimental protocols on the experiements page.

Project Design
Transformation of the induction systems
The induction systems were linked to both the thioesterases and fluorescent proteins for proof of concept. In total, six different plasmids were designed:



We used three different thioesterases for stopping the elongation of FAs at different target lengths, two bacterial thioesterases (TesBT and TesA) and one plant thioesterase (FatB). We subsequently utilized three induction systems, consisting of inducible promoter sequences, for tuning the expression of each thioesterase gene. Each induction system reacts to its corresponding stimuli:
- CUP: Inducible by addition of copper ions.
- TetON: Inducible by addition of tetracycline (or one of its derivatives).
- EstrInd: Inducible by addition of estradiole.
In addition to the three plasmids harboring each thioesterase-linked induction system, we also constructed three plasmids for benchmarking the response curves for each inducer. These plasmids consist of each of induction systems coupled to the expression of three different fluorescent proteins (RFP, BFP and GFP). Once these benchmarking plasmids containing the fluorescent proteins were constructed and integrated in yeast, we subsequently measured the expression levels of the fluorophores across a wide range of inducer concentrations using flow cytometry.

Several different plasmids were used as backbone in the design. These contain different yeast selection markers, enabling simultaneous integration and cultivation of yeast. Backbone plasmid p413TEF was used for the CUP based systems. It contains the His3 selection marker for yeast and an ampicillin resistance gene for E. coli cultivation. p415TEF was used for the TetON induction systems, this plasmid contains the Leu2 selection marker for yeast and an ampicillin resistance gene for E. coli cultivation. Backbone plasmid p416TEF was used for the plasmid containing the EstrInd-FatB system, while p416GFP is used for the GFP and EstrInd linked induction system. Both p416TEF and p416GFP contain the Ura3 selection marker and an ampicillin resistance gene.
The last part of the design process before entering the physical lab, involves creating primers containing a 20bp overhang in each end, permitting Gibson assembly of the different ordered gene fragments into plasmids. Primers and thioesterase gene fragments were ordered from IDT and Eurofins. Two of the induction systems, EstrInd and TetON, were kindly provided to us by the Tom Ellis Lab at Imperial College Centre for Synthetic Biology (IC-CSynB) and the Department of Bioengineering at Imperial College, London. All other gene fragments were fortunately provided by the Systems and Synthetic Biology division at Chalmers University of Technology. Our supervisor Andrea, helped us assembly the VP16-Estradiole-zif268 gene fragement for the EstrInd system together with promotor (ScTEF1p) and terminator (ScADH1 T) using MoClo assemblies. The same applies for the TetA-NLS-Gal4AD for the TetON system, which was assembled together with promotor (ScTEF1p) and terminator (ScPGK1 T).
We used PCR amplification for the gene fragments and digested the backbone plasmids using restriction enzymes in preparation for Gibson Assembly Cloning, with one exception. For p416GFP we used PCR-mediated deletion of the non-target sequence to retain the GFP gene in the backbone. In order to verify digestions and successful PCRs we used gel electrophoresis.
The last part of the design process before entering the physical lab, involves creating primers containing a 20bp overhang in each end, permitting Gibson assembly of the different ordered gene fragments into plasmids. Primers and thioesterase gene fragments were ordered from IDT and Eurofins. Two of the induction systems, EstrInd and TetON, were kindly provided to us by the Tom Ellis Lab at Imperial College Centre for Synthetic Biology (IC-CSynB) and the Department of Bioengineering at Imperial College, London. All other gene fragments were fortunately provided by the Systems and Synthetic Biology division at Chalmers University of Technology. Our supervisor Andrea, helped us assembly the VP16-Estradiole-zif268 gene fragement for the EstrInd system together with promotor (ScTEF1p) and terminator (ScADH1 T) using MoClo assemblies. The same applies for the TetA-NLS-Gal4AD for the TetON system, which was assembled together with promotor (ScTEF1p) and terminator (ScPGK1 T).
We used PCR amplification for the gene fragments and digested the backbone plasmids using restriction enzymes in preparation for Gibson Assembly Cloning, with one exception. For p416GFP we used PCR-mediated deletion of the non-target sequence to retain the GFP gene in the backbone. In order to verify digestions and successful PCRs we used gel electrophoresis.

The gene fragments were then Gibson assembled into plasmids followed by transformation into chemically competent E. coli. E. coli was cultured on ampicillin LB plates and media and plasmids were harvested from after successful transformation. To verify if the assembly was successful, we performed diagnostic digests and gel electrophoresis on the harvested plasmids. Plasmids with correct lengths were also verified using Sanger sequencing.
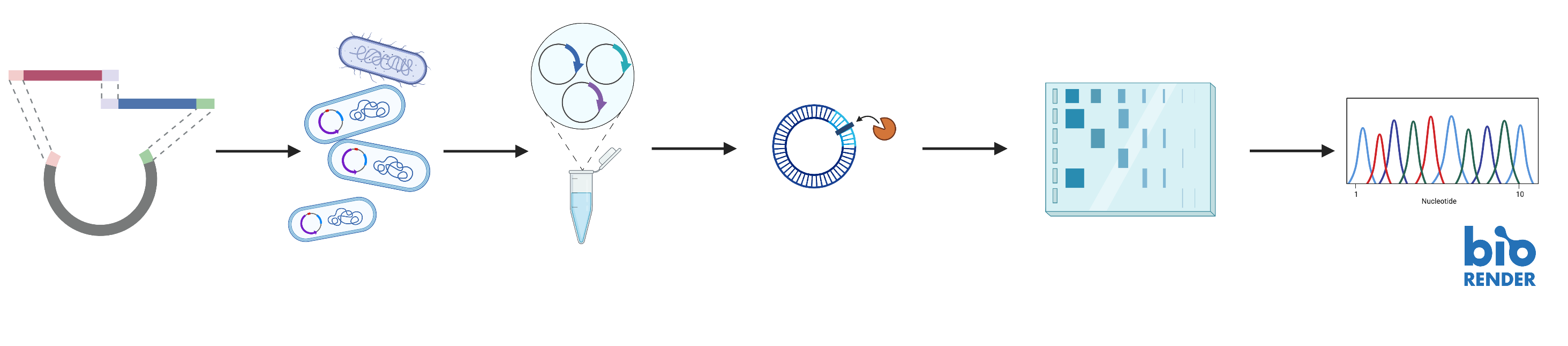
Finally, correctly assembled plasmids were transformed into the S. Cerevisiae strain CEN.PK 102-5B. Yeast containing each of the different combinations of induction plasmids were created. Using selective media lacking corresponding combinations of the selective factors: Ura3, Leu2 and His3 for each respective yeast transformation, we ensured that we only have cells containing the correct plasmid combinations.

Integration of bacterial eFAS system
Since we needed to integrate eight bacterial genes (acpP, acpS, fabB, fabG, fabZ, fabI, fabD, fabH) to our yeast through transformation within the limited timeframe of the project. To speed up the process, we designed our integration fragments to include two genes in each fragment. This meant we needed to perform four genome integrations instead of eight. We used a CRISPR/Cas9 based marker free system to introduce our gene fragments into the genome.
We codon optimised all gene sequences for S. cerevisiae since they come from multiple other species. Since the marker free plasmids contain two terminators, we can insert the gene fragments in between the terminators with the genes facing opposite directions. The designed fragment consists of two different promoters facing opposite directions followed by two different genes. Different promoters were used to avoid unwanted homologous recombination.
We codon optimised all gene sequences for S. cerevisiae since they come from multiple other species. Since the marker free plasmids contain two terminators, we can insert the gene fragments in between the terminators with the genes facing opposite directions. The designed fragment consists of two different promoters facing opposite directions followed by two different genes. Different promoters were used to avoid unwanted homologous recombination.

The gene fragments are then inserted into four different marker free plasmids, pCfB3038, pCfB3040, pCfB2909 and pCfB3039. pCfB3038 integrates at site XII-1 in chromosome XII, pCfB3040 integrates at site XII-4 in chromosome XII, pCfB2909 integrates at site XII-5 in chromosome XII and pCfB3038 integrates at site XII-2 in chromosome XII. The plasmids were propagated in E. coli and purified in preparation for assembly.



The plasmids were digested with enzymes to be linearized and PCR was performed on each of the gene fragments with primers containing overhangs matching the digested plasmid sticky ends. The PCR products and digested plasmids were verified using gel electrophoresis and gel purified when needed. Gibson assembly was then performed to assembley the gene fragments together with the plasmid.



The plasmids were then propagated in E. coli, purified, digested, verified with gel electrophoresis, and sequenced to ensure correct assembly before transformation into yeast.


The assembled plasmids were transformed into yeast one by one together with a plasmid containing cas9 plasmid and gRNA corresponding to the specific integration site. The plasmid containing the gene fragment was digested before being transformed into the yeast, since the linear fragment will be integrated into the yeast genome at the specific integration site.
The gRNA/cas9 plasmids contain selection markers, allowing the yeast to survive plating on a YPD plate containing the specific selection antibiotic. Potential transformants harbouring the integrated genes were then picked, boiled to lyse the cells, and verified using PCR with primers that bind to either side of the integration site. The PCR products were run on a gel and sequenced to verify that the fragments were integrated correctly.
The gRNA/cas9 plasmids contain selection markers, allowing the yeast to survive plating on a YPD plate containing the specific selection antibiotic. Potential transformants harbouring the integrated genes were then picked, boiled to lyse the cells, and verified using PCR with primers that bind to either side of the integration site. The PCR products were run on a gel and sequenced to verify that the fragments were integrated correctly.
Antisense RNA
When introducing a new E. coli FAS system into our yeast we realised that we would have to downregulate the native FAS system in the yeast, therefore we designed an antisense RNA system as part of our engineering cycle.
The antisense system was designed to downregulate two genes, one is for downregulating the wild type of strain and one for the high fatty acid producing yeast, meaning we could use the same gene fragment to downregulate the native FAS system in both types. It is assembled, verified and transformed in the same way as the other four gene fragments described earlier. The antisense gene fragment is inserted into a marker free plasmid, pCfB3035. This plasmid corresponds to integration site X-4 in chromosome X.
The antisense system was designed to downregulate two genes, one is for downregulating the wild type of strain and one for the high fatty acid producing yeast, meaning we could use the same gene fragment to downregulate the native FAS system in both types. It is assembled, verified and transformed in the same way as the other four gene fragments described earlier. The antisense gene fragment is inserted into a marker free plasmid, pCfB3035. This plasmid corresponds to integration site X-4 in chromosome X.
